 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 47(3); 2013 > Article
-
Case Study
Imprint Cytology of Soft Tissue Myoepithelioma: A Case Study - Seok Ju Park, Ae Ri Kim, Mi Jin Gu, Joon Hyuk Choi, Duk Seop Shin1
-
Korean Journal of Pathology 2013;47(3):299-303.
DOI: https://doi.org/10.4132/KoreanJPathol.2013.47.3.299
Published online: June 25, 2013
Department of Pathology, Yeungnam University College of Medicine, Daegu, Korea.
1Department of Orthopedic Surgery, Yeungnam University College of Medicine, Daegu, Korea.
- Corresponding Author: Joon Hyuk Choi, M.D. Departments of Pathology, Yeungnam University College of Medicine, 170 Hyeonchung-ro, Nam-gu, Daegu 705-717, Korea. Tel: +82-53-620-3335, Fax: +82-53-656-1429, joonhyukchoi@ynu.ac.kr
• Received: November 16, 2012 • Revised: December 27, 2012 • Accepted: January 10, 2013
© 2013 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- Soft tissue myoepithelioma is a rare neoplasm composed of myoepithelial cells. Here, we describe the cytologic features of soft tissue myoepithelioma arising on the right forearm in an 18-year-old man. The excised tumor (3.0×1.8×1.5 cm) was well-demarcated, yellow-gray, soft, and myxoid. The cytologic smears showed round to spindle, epithelioid, and plasmacytoid cells in the myxoid background. The nuclei were uniform, round to ovoid, with finely distributed chromatin and eosinophilic or pale cytoplasm. The tumor cells demonstrated immunoreactivity for cytokeratin (AE1/AE3), epithelial membrane antigen, S100 protein, and glial fibrillary acidic protein. Electron microscopy showed intermediate filaments, desmosomes, and basal lamina.
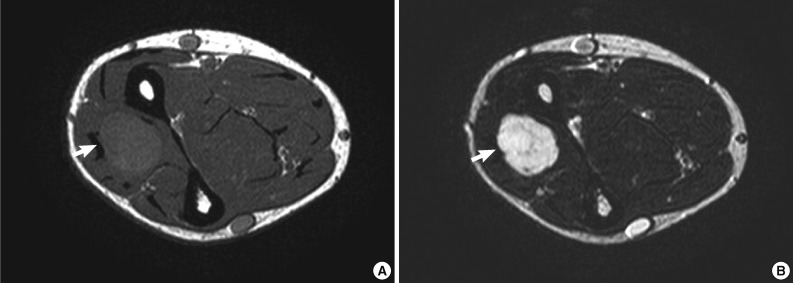
- An 18-year-old man presented with a soft tissue mass of five months' duration in the right forearm. There was no trauma history. Physical examination revealed a round, tender mass. On magnetic resonance scan of the right forearm, T1-weighted imaging revealed a circumscribed mass with intermediate signal intensity. T2-weighted imaging revealed high signal intensity within the mass (Fig. 1A, B). Gadolinium-enhanced fat-suppressed T1-weighted imaging of the mass showed inhomogeneous enhancement. There was no bony involvement. The initial clinical impression was schwannoma. The mass was excised and imprint cytology of the mass was performed.
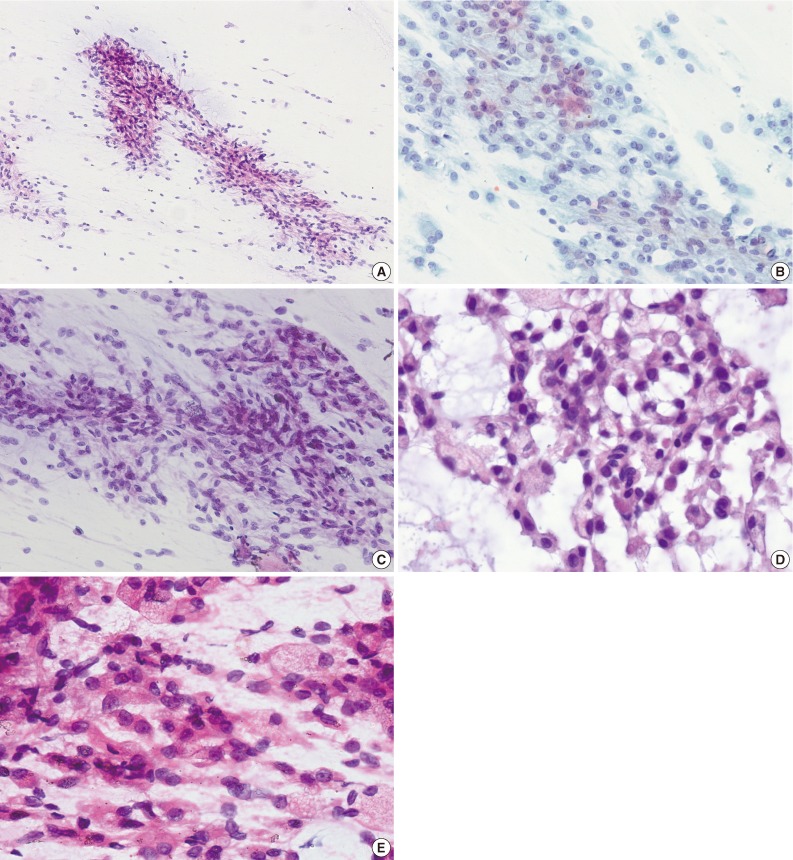
- The obtained slides were stained with hematoxylin and eosin and Papanicolaou methods. On microscopic examination, the smears were moderately cellular. The smear revealed clusters, sheets, and isolated cells in the background of myxoid materials (Fig. 2A-E). Tissue fragments were occasionally scattered. The cells were round to spindle and epithelioid-shaped. The nuclei were uniformly round to ovoid, with finely distributed chromatin and a small nucleolus. The nuclear membrane was smooth. The cytoplasmic borders were well-defined or indistinct. The tumor cells had abundant eosinophilic or pale cytoplasm. Some cells had eccentrically-located nuclei and lesser amount of basophilic cytoplasm, resulting in a plasmacytoid appearance. Long, tapering cytoplasmic processes were also seen. Cytoplasmic vacuolation was noted in some tumor cells. Cells with naked nuclei were also present. Fibrillary materials were occasionally seen in the background. No nuclear pleomorphism was present. Neither mitosis nor necrosis was found. On immunohistochemical staining for cytologic slides, the tumor cells were occasionally positive for cytokeratin (AE1/AE3) and S100 protein.
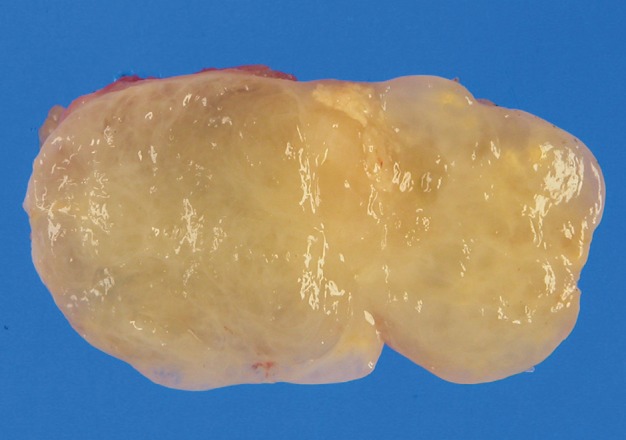
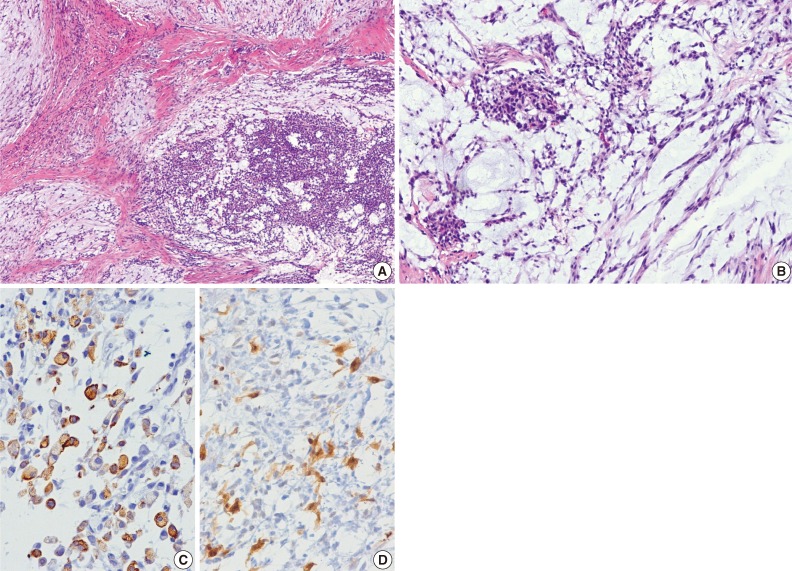
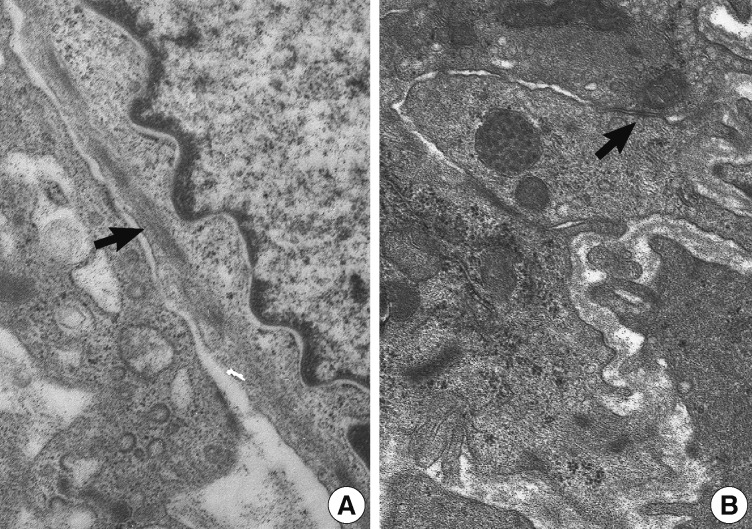
- The excised tumor measured 3.0×1.8×1.5 cm. The tumor was well-demarcated, yellow solid, soft, and myxoid (Fig. 3). Histologically, the tumor showed multilobulated growth with myxoid and fibrous stroma. The tumor consisted of epithelioid, spindle and plasmacytoid cells (Fig. 4A, B). The tumor cells were present in solid sheets or reticular pattern without ductal component or necrosis. The tumor cells showed immunostaining for cytokeratin (AE1/AE3), epithelial membrane antigen (EMA), S100 protein, and glial fibrillary acidic protein (GFAP) (Fig. 4C, D). Electron microscopic examination showed intermediate filaments, desmosomes, and basal lamina (Fig. 5). The histologic features were consistent with soft tissue myoepithelioma. Neither recurrence nor metastasis was noted four year follow-up.
CASE REPORT
- Soft tissue myoepithelioma is a rare tumor composed of neoplastic cells with features of myoepithelial differentiation. The tumor presents as a painless or painful mass in the dermis, subcutis, or deep soft tissue.3,4 While most myoepitheliomas are of salivary gland origin, they have been reported in the soft tissue, retroperitoneum,3 lung,5 and testis.6 The histogenesis of myoepithelial tumors arising in soft tissue is unknown. It likely reflects a different pattern of gene expression during oncogenesis rather than origin of a specific cell lineage.7 Hallor et al.8 reported that a minimally deleted region of 3.55 Mb at chromosome band 19p13 was identified in soft tissue myoepitheliomas. In the present case, the tumor consisted of epithelioid, spindle, and plasmacytoid cells forming solid sheets or reticular pattern in myxoid and fibrous stroma. The tumor cells revealed positive reaction for cytokeratin (AE1/AE3), EMA, S100 protein, and GFAP. The cytomorphologic and immunohistochemical features of the present case are similar to those of myoepitheliomas arising in the salivary gland.9-12
- Soft tissue myoepitheliomas, mixed tumors, and parachordomas are on the spectrum of tumor showing myoepithelial differentiation.1 Characteristically, mixed tumor has more pronounced ductal differentiation, while parachordoma shows prominent cytoplasmic vacuolation. In the present case, the tumor had no ductal component and no cytoplasmic vacuolation. Ultrastructurally, myoepitheliomas show intermediate filaments, desmosomes, and basal lamina.13 Intermediate filaments, desmosomes, and basal lamina were present in this case.
- Soft tissue myoepitheliomas may be mistaken as other type of soft tissue tumors due to their cytomorphologically heterogeneous features. The differential diagnosis of soft tissue myoepithelioma includes mixed tumor, parachrodoma, schwannoma, smooth muscle tumor, ossifying fibromyxoid tumor (OFMT), myxoid liposarcoma, extraskeletal myxoid chondrosarcoma (EMC), and metastatic carcinoma. Mixed tumor of soft tissue is uncommon. It shows epithelial and myoepithelial cells in chondromyxoid background. In addition, acinar or duct-like arrangement usually present. Parachordoma shows spindle to epithelioid cells, with clear, vacuolated cytoplasm. Immunohistochemically, parachordomas are positive for cytokeratin and S100 protein.14 Schwannoma has wavy, point-ended nuclei in collagenous or myxoid background and nuclear palisading.15 Smooth muscle tumor has cigar-shaped, blunt-ended nuclei and eosinophilic fibrillary cytoplasm.16 Conversely, the cells of soft tissue myoepithlioma have more tapered nuclei. Immunoreactivity for smooth muscle actin, desmin, and h-caldesmon supports a diagnosis of smooth muscle tumor. OFMT shows round and ovoid cells in myxoid matrix.17 OFMT is immunoreactive for S100 protein in approximately 70% of cases but negative for cytokeratin and GFAP. Myxoid liposarcoma shows lipoblasts and delicate, arborizing, thin-walled blood vessels in myxoid background.18 EMC is characterized by spindle, stellate, or round cells in a myxoid or chondromyxoid matrix and shows variable immunoreactivity for S100 protein, neuron-specific enolase, and synaptophysin.19,20 Generally, metastatic carcinomas show epithelial tumor cells with hyperchromatic nuclei, prominent nucleoli, and a high nuclear to cytoplasmic ratio. It can be excluded by the absence of immunoreactivity for S100 protein and myogenic markers. Immunohistochemical expression for epithelial markers (cytokeratin and/or EMA), S100 protein or GFAP is useful for confirmation of myoepithelial differentiation.3,7
- Although most cases of myoepithelial neoplasms of soft tissue are benign, approximate 20% have a risk for local recurrence.3 The histopathologic criteria for malignancy in soft tissue myoepithelial neoplasms are moderate to severe cytologic atypia, increase of nuclear to cytoplasmic ratio, nuclear pleomorphism, and readily identifiable nucleoli.2,3 No cytologic features of malignancy were present in this case of myoepithelioma. The treatment is complete surgical excision with a clear margin.
- In conclusion, recognition of the cytomorphologic features of soft tissue myoepithelioma is necessary for the correct cytological diagnosis. Soft tissue myoepithelioma should be included in the differential diagnosis of soft tissue epithelioid and spindle cell neoplasms.
DISCUSSION
- 1. Kilpatrick SE, Limon J. Mixed tumour/myoepithelioma/parachordoma. In: Fletcher CD, Unni K, Mertens F, eds. WHO classification of tumours: pathology and genetics of tumours of soft tissue and bone. Lyon: IARC Press, 2002; 198-199.
- 2. Kilpatrick SE, Hitchcock MG, Kraus MD, Calonje E, Fletcher CD. Mixed tumors and myoepitheliomas of soft tissue: a clinicopathologic study of 19 cases with a unifying concept. Am J Surg Pathol 1997; 21: 13-22. PubMed
- 3. Hornick JL, Fletcher CD. Myoepithelial tumors of soft tissue: a clinicopathologic and immunohistochemical study of 101 cases with evaluation of prognostic parameters. Am J Surg Pathol 2003; 27: 1183-1196. PubMed
- 4. Hornick JL, Fletcher CD. Cutaneous myoepithelioma: a clinicopathologic and immunohistochemical study of 14 cases. Hum Pathol 2004; 35: 14-24. ArticlePubMed
- 5. Veeramachaneni R, Gulick J, Halldorsson AO, Van TT, Zhang PL, Herrera GA. Benign myoepithelioma of the lung: a case report and review of the literature. Arch Pathol Lab Med 2001; 125: 1494-1496. PubMed
- 6. Jeong SM, Lee JH, Park WY, et al. Primary myoepithelioma of the testis: a case report. Korean J Pathol 2011; 45(Suppl 1): S20-S24. Article
- 7. Gleason BC, Fletcher CD. Myoepithelial carcinoma of soft tissue in children: an aggressive neoplasm analyzed in a series of 29 cases. Am J Surg Pathol 2007; 31: 1813-1824. ArticlePubMed
- 8. Hallor KH, Teixeira MR, Fletcher CD, et al. Heterogeneous genetic profiles in soft tissue myoepitheliomas. Mod Pathol 2008; 21: 1311-1319. ArticlePubMedPDF
- 9. Suh CH, Chun HJ. Plasmacytoid myoepithelioma of the parotid gland: a case report with ultrastructural and immunohistochemical findings. Korean J Pathol 1988; 22: 324-330.
- 10. Kumar PV, Sobhani SA, Monabati A, Hashemi SB, Eghtadari F, Hamidi SA. Myoepithelioma of the salivary glands: fine needle aspiration biopsy findings. Acta Cytol 2004; 48: 302-308. PubMed
- 11. Das DK, Haji BE, Ahmed MS, Hossain MN. Myoepithelioma of the parotid gland initially diagnosed by fine needle aspiration cytology and immunocytochemistry: a case report. Acta Cytol 2005; 49: 65-70. PubMed
- 12. Kim NR, Cho HY, Ha SY. Cytology of plasmacytoid type myoepithelioma: report of two cases. Korean J Pathol 2009; 43: 489-493. Article
- 13. Bisceglia M, Cardone M, Fantasia L, Cenacchi G, Pasquinelli G. Mixed tumors, myoepitheliomas, and oncocytomas of the soft tissues are likely members of the same family: a clinicopathologic and ultrastructural study. Ultrastruct Pathol 2001; 25: 399-418. ArticlePubMed
- 14. Folpe AL, Agoff SN, Willis J, Weiss SW. Parachordoma is immunohistochemically and cytogenetically distinct from axial chordoma and extraskeletal myxoid chondrosarcoma. Am J Surg Pathol 1999; 23: 1059-1067. ArticlePubMed
- 15. Domanski HA, Akerman M, Engellau J, Gustafson P, Mertens F, Rydholm A. Fine-needle aspiration of neurilemoma (schwannoma): a clinicocytopathologic study of 116 patients. Diagn Cytopathol 2006; 34: 403-412. ArticlePubMed
- 16. Barbazza R, Chiarelli S, Quintarelli GF, Manconi R. Role of fine-needle aspiration cytology in the preoperative evaluation of smooth muscle tumors. Diagn Cytopathol 1997; 16: 326-330. ArticlePubMed
- 17. Mohanty SK, Srinivasan R, Rajwanshi A, Vasishta RK, Vignesh PS. Cytologic diagnosis of ossifying fibromyxoid tumor of soft tissue: a case report. Diagn Cytopathol 2004; 30: 41-45. ArticlePubMed
- 18. Pantanowitz L, Otis CN. Myxoid liposarcoma. Diagn Cytopathol 2007; 35: 283-284. ArticlePubMed
- 19. Wadhwa N, Arora VK, Singh N, Bhatia A. Fine needle aspiration cytology of primary extraskeletal myxoid chondrosarcoma: a case report. Acta Cytol 2000; 44: 445-448. PubMed
- 20. Handa U, Singhal N, Punia RS, Garg S, Mohan H. Cytologic features and differential diagnosis in a case of extraskeletal mesenchymal chondrosarcoma: a case report. Acta Cytol 2009; 53: 704-706. PubMed
REFERENCES
Fig. 1Magnetic resonance images of the right forearm. (A) An intermediate signal mass is present in the right forearm on axial T1-weighted image (arrow). (B) The mass shows a high signal intensity on axial T2-weighted image (arrow).
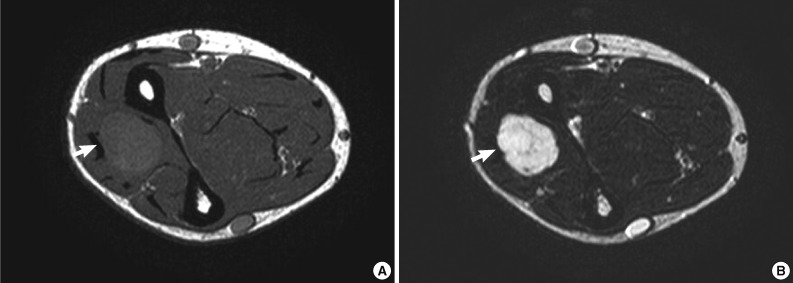
Fig. 2Cytologic findings. (A) The smears are moderately cellular. The tumor cells are present in clusters, sheets and isolated cells in myxoid background. (B) The tumor cells have uniformly round and oval nuclei with finely distributed chromatin and a small nucleolus (Papanicolaou stain). (C) Spindle cells are arranged in clusters. (D) The cells have eccentrically displaced nuclei with basophilic or vacuolated cytoplasm. (E) Epithelioid cells with abundant eosinophilic cytoplasm are present.
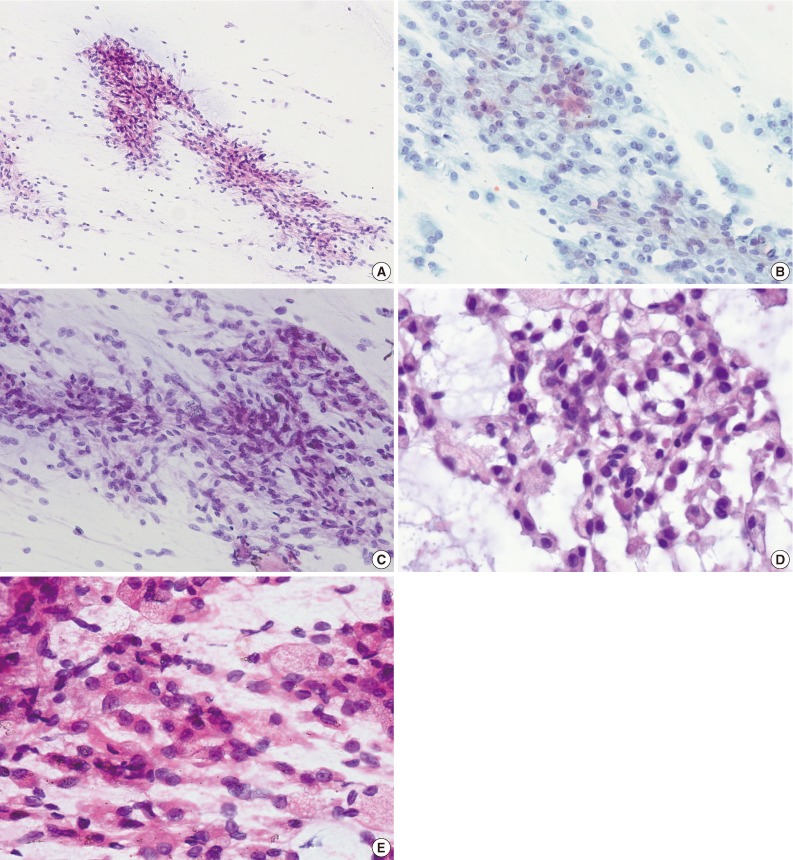
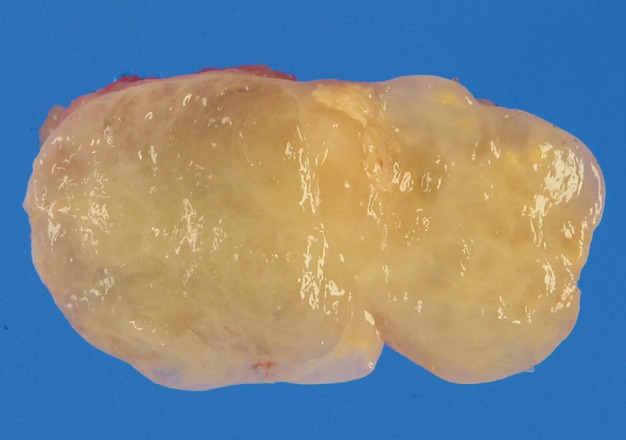
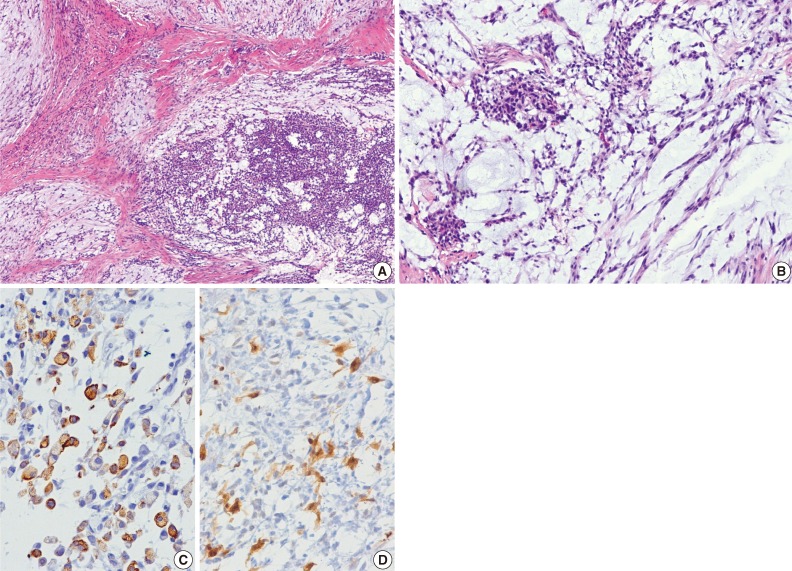
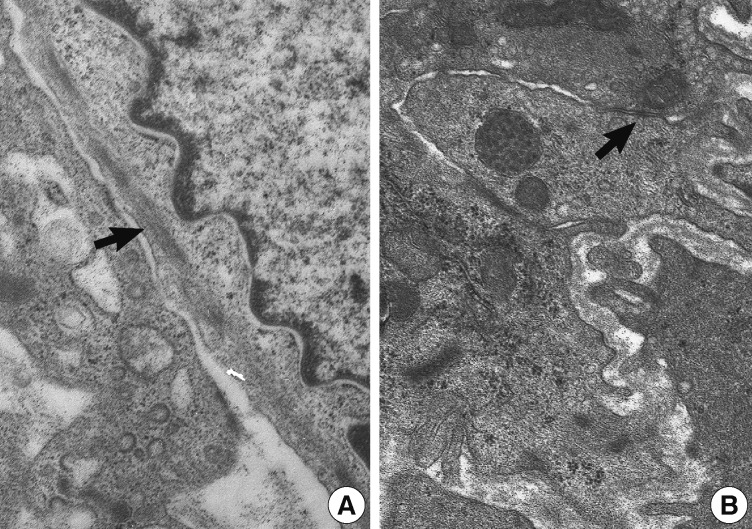
Figure & Data
References
Citations
Citations to this article as recorded by 
- Myoepithelial tumors of soft tissue and bone in children and young adults: A clinicopathologic study of 40 cases occurring in patients ≤ 21 Years of age
Suzanna J. Logan, Carina A. Dehner, Fatimah I. Alruwaii, Nasir Ud Din, Damon R. Olson, Karen J. Fritchie, Gregory W. Charville, Melissa M. Blessing, Andrew L. Folpe
Human Pathology.2024; 149: 10. CrossRef - Fine-needle aspiration cytopathology of soft tissue myoepithelioma: an analysis of seven cases
Paul E. Wakely, Momin T. Siddiqui
Journal of the American Society of Cytopathology.2022; 11(1): 31. CrossRef - Cytology‐histology correlation of myoepithelial tumors harboring EWSR1‐POU5F1 fusions: A report of two cases
Ian A. Gelarden, Lucy Fu, Kai Lee Yap, Aida I. Richardson, Pauline M. Chou
Diagnostic Cytopathology.2022;[Epub] CrossRef - A case of myoepithelial carcinoma of the left shoulder
Shuhei ISHII, Noriyuki FURUTA, Kyoko KOMATSU, Yoshiya SUGIURA, Noriko MOTOI, Yutaka TAKAZAWA, Yuko SUGIYAMA, Yuichi ISHIKAWA
The Journal of the Japanese Society of Clinical Cytology.2018; 57(2): 129. CrossRef - Fine‐needle aspiration of soft tissue myoepithelioma
Gang Wang, Tracy Tucker, Tony L. Ng, Carlos F. Villamil, Malcolm M. Hayes
Diagnostic Cytopathology.2016; 44(2): 152. CrossRef - A case report of spindle cell myoepithelioma with extensive lipomatous metaplasia and thick collagen bundles in the submandibular gland
Mi Jung Kwon, Hye Jeong Kim, Bumjung Park, Seong Jin Cho, Hyung Sik Shin, Hye‐Rim Park, Soo Kee Min, Jinwon Seo, Kyueng‐Whan Min, Eun Sook Nam
Diagnostic Cytopathology.2016; 44(9): 764. CrossRef - Myoepithelioma of soft tissue, a case report
Hassania Ameurtesse, Leila Chbani, JM Coindre, Hinde Elfatemi, Toufik Harmouch, Afaf Amarti
Research.2014;[Epub] CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
Imprint Cytology of Soft Tissue Myoepithelioma: A Case Study





Fig. 1 Magnetic resonance images of the right forearm. (A) An intermediate signal mass is present in the right forearm on axial T1-weighted image (arrow). (B) The mass shows a high signal intensity on axial T2-weighted image (arrow).
Fig. 2 Cytologic findings. (A) The smears are moderately cellular. The tumor cells are present in clusters, sheets and isolated cells in myxoid background. (B) The tumor cells have uniformly round and oval nuclei with finely distributed chromatin and a small nucleolus (Papanicolaou stain). (C) Spindle cells are arranged in clusters. (D) The cells have eccentrically displaced nuclei with basophilic or vacuolated cytoplasm. (E) Epithelioid cells with abundant eosinophilic cytoplasm are present.
Fig. 3 Gross findings. The tumor is well-circumscribed, yellow-gray, soft, and myxoid.
Fig. 4 Histologic findings. (A) At low power, the tumor shows lobular architecture in myxoid and fibrous stroma. (B) The tumor cells are arranged in reticular pattern and solid nests. (C, D) The tumor cells show positivity for cytokeratin (AE1/AE3) (C) and S100 protein (D).
Fig. 5 Ultrastructural findings. Intermediate filaments (A, arrow) and desmosomes (B, arrow) are present (A, ×9,000; B, ×10,000).
Fig. 1
Fig. 2
Fig. 3
Fig. 4
Fig. 5
Imprint Cytology of Soft Tissue Myoepithelioma: A Case Study

