 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 48(4); 2014 > Article
-
Brief Case Report
Perivascular Epithelioid Cell Tumor Arising in the Sacrum: A Case Report - Yoon Sung Bae, Se Hoon Kim
-
Korean Journal of Pathology 2014;48(4):331-334.
DOI: https://doi.org/10.4132/KoreanJPathol.2014.48.4.331
Published online: August 26, 2014
Department of Pathology, Yonsei University College of Medicine, Seoul, Korea.
- Corresponding Author: Se Hoon Kim, M.D. Department of Pathology, Yonsei University College of Medicine, 50 Yonsei-ro, Seodaemun-gu, Seoul 120-752, Korea. Tel: +82-2-2228-1769, Fax: +82-2-362-0860, paxco@yuhs.ac
• Received: August 21, 2013 • Revised: September 30, 2013 • Accepted: October 1, 2013
© 2014 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 8,564 Views
- 45 Download
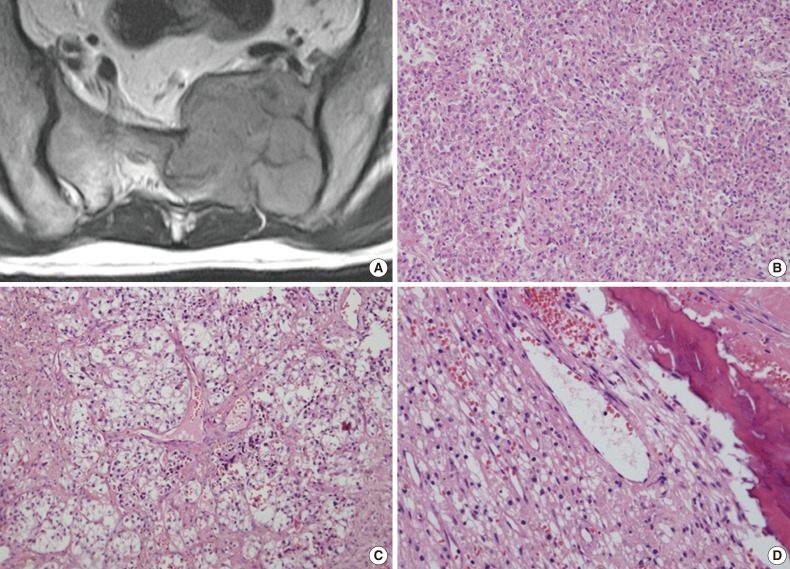
- A 45-year-old man without relevant medical history presented with left buttock and leg pain 1.5 months in duration. Sacral magnetic resonance imaging revealed a 5.5×4.7-cm lobulated osteolytic expansile mass on the left side of the sacrum (Fig. 1A). The tumor was invading the lumbo-sacral spinal canal medially via neural foramens S1-3 and laterally via the left sacroiliac joint. Positron emission tomography showed increased fluorodeoxyglucose uptake, suggesting malignancy. Due to the radiologic characteristics, a germ cell tumor or nerve sheath tumor was considered. An excisional surgery was performed, and intraoperatively the tumor was found to be solid and multilobulated, and was covered with a thin vascularized capsule. It was easily separated from the sacral nerve roots and sacral bone, thus allowing for near total removal. The tumor bed was then treated with postoperative radiation therapy (64.8 Gy) without complication.
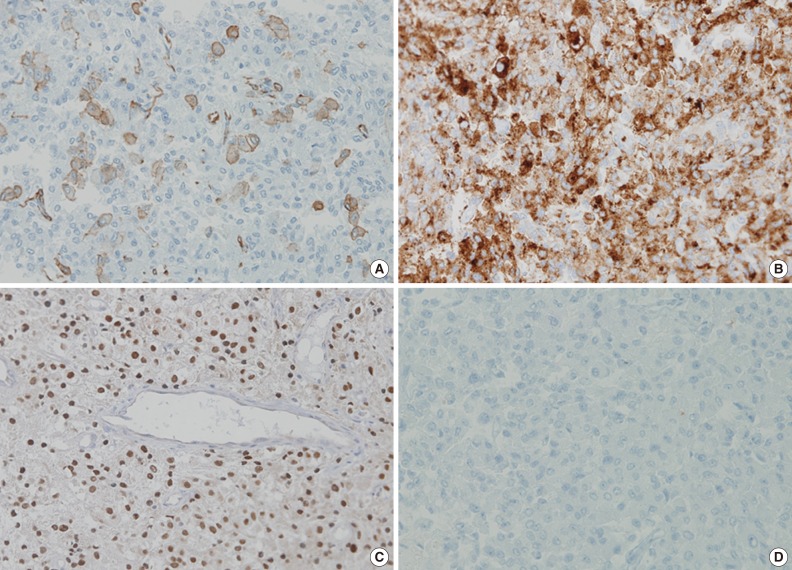
- The tumor was relatively poorly demarcated from normal tissue and showed rich networks of arborizing sinusoidal capillaries. It was composed exclusively of polygonal and epithelioid cells with abundant clear to eosinophilic cytoplasm, most of which was arranged in sheets or a vague nesting pattern (Fig. 1B). Occasional perivascular arrangements were also noted (Fig. 1C). Infiltration into adjacent bone tissue was found (Fig. 1D), but no necrosis or mitosis was observed. The initial differential diagnosis included ependymoma, hemangioblastoma, paraganglioma, and epithelioid neurogenic tumor. Many IHC stains were performed, but none of the tumor cells showed immunoreactivity to antibodies except smooth muscle actin (SMA) (Fig. 2A). Several additional IHC stains were done and human melanoma black 45 (HMB45) and transcription factor E3 (TFE3) were found to be expressed by the tumor cells (Fig. 2B, C), but the tumor was negative for Melan A (Fig. 2D). With these IHC results, a confirmative diagnosis of PEComa was established.
CASE REPORT
- PEComa is an uncommon tumor that can occur in various organs, with the uterus being a representative primary site of PEComa. Angiomyolipoma of the kidney and liver, and clear cell tumor of the lung are also regarded to originate from perivascular epithelioid cells. When this tumor arises in other organs it can be difficult to diagnose. In this case, PEComa was not included in the initial differential diagnosis. Moreover, perivascular arrangements, the most unique characteristic of a PEComa, were not prominent in this case because they were masked by the diffuse and sheet-like growth patterns of tumor cells. Initial IHC stains were not informative, so additional stains were performed which allowed for the eventual diagnosis of PEComa.
- Diagnosis of PEComa is quite challenging and requires a combination of attention to morphologic detail and the appropriate use of melanocytic and myogenic markers, such as HMB45, Melan A, and SMA. TFE3, a melanocytic marker, can also be expressed in PEComa. However, malignant melanoma (MM) and clear cell sarcoma (CCS) can exhibit a similar IHC profile, which remains a diagnostic dilemma. Distinguishing between these tumors is important because the majority of PEComas follow a benign clinical course in contrast to the poor prognosis of MM and CCS, although rare malignant PEComas do occur.2 MM can have similar morphological patterns to PEComa and shares IHC profiles including HMB45, Melan A, and TFE3. Furthermore, SMA, a marker generally negative in melanoma, has been reported to be expressed in a desmoplastic variant of MM.5 S100 has been proposed as a useful marker because it is expressed in MM, but not in PEComa, even though a small subset of PEComas was found to express it.2 Multiple myeloma 1 (MUM1), a B-cell proliferation marker, also can be used because of its expression in the majority of MMs in contrast to the significantly diminished or absent expression seen in PEComa.6,7 Therefore, the addition of S100 protein and MUM1 to IHC panels could be helpful in the differential diagnosis between these lesions. In our case, the tumor cells were positive for SMA and negative for S100 protein and MUM1, which supported a diagnosis of PEComa rather than of MM. CCS can also mimic PEComa morphologically and express melanocytic markers. However, CCS can be distinguished from PEComa because SMA is consistently negative in CCS.
- This case is noteworthy in that the tumor cells of a PEComa exhibited TFE3 expression. TFE3 is a member of microphthalmia transcription factor family and has been associated with several neoplastic conditions including alveolar soft part sarcoma (ASPS), Xp11 translocation renal cell carcinoma and PEComa.8,9 The likelihood of ASPS was considered to be low in this case because the cell nuclei were less atypical than those usually seen in ASPS, no crystalloid material was detected on periodic acid-Schiff stain and HMB45 was clearly positive.2 Recently, Argani et al.10 presented distinctive features of TFE3-positive PEComa, which included relatively young patient age, predominant nesting architecture with epithelioid cytology, lack of nuclear anaplasia or pleomorphism, weak positivity to IHC stains for smooth muscle markers and absence of a tuberous sclerosis complex. Our case shares several clinicopathologic aspects with previously reported cases.
- Although most PEComas have a benign clinical course, several cases with local invasion, recurrence and even distant metastasis have been reported.2 At this point, the biologic behavior of PEComa is unclear. Recently, Folpe et al.2 proposed criteria for estimating the malignant potential of PEComa. According to that report, tumors with large size (more than 5 cm), infiltrative margins, nuclear pleomorphism, mitosis, and necrosis may be more aggressive. In this case, the tumor consisted of monotonous tumor cells without nuclear atypia, mitosis or necrosis. However, it was regarded as at least a borderline or low-grade malignancy because it was larger than 5 cm and had an ill-defined tumor border.
- The significance of this case is the rare site of occurrence. Although this disease can occur in a wide range of anatomic sites, bone as a primary site of a PEComa is very unusual. There have been several reports concerning PEComas arising from skeletal tissue, including the skull, rib and fibula. The sacrum, however, has never been reported as a primary site of a PEComa, and ours is the first reported case of a PEComa arising in this location. Diagnosing PEComas at unusual sites can be challenging for pathologists, particularly if this disease is not suspected in the initial differential diagnosis.
DISCUSSION
- 1. Martignoni G, Pea M, Reghellin D, Zamboni G, Bonetti F. PEComas: the past, the present and the future. Virchows Arch 2008; 452: 119-132. ArticlePubMedPDF
- 2. Folpe AL, Mentzel T, Lehr HA, Fisher C, Balzer BL, Weiss SW. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: a clinicopathologic study of 26 cases and review of the literature. Am J Surg Pathol 2005; 29: 1558-1575. PubMed
- 3. Hornick JL, Fletcher CD. PEComa: what do we know so far? Histopathology 2006; 48: 75-82. ArticlePubMed
- 4. Yamashita K, Fletcher CD. PEComa presenting in bone: clinicopathologic analysis of 6 cases and literature review. Am J Surg Pathol 2010; 34: 1622-1629. ArticlePubMed
- 5. Carlson JA, Dickersin GR, Sober AJ, Barnhill RL. Desmoplastic neurotropic melanoma: a clinicopathologic analysis of 28 cases. Cancer 1995; 75: 478-494. ArticlePubMed
- 6. Ferenczi K, Lastra RR, Farkas T, et al. MUM-1 expression differentiates tumors in the PEComa family from clear cell sarcoma and melanoma. Int J Surg Pathol 2012; 20: 29-36. ArticlePubMedPDF
- 7. Sundram U, Harvell JD, Rouse RV, Natkunam Y. Expression of the B-cell proliferation marker MUM1 by melanocytic lesions and comparison with S100, gp100 (HMB45), and MelanA. Mod Pathol 2003; 16: 802-810. ArticlePubMed
- 8. Argani P, Lal P, Hutchinson B, Lui MY, Reuter VE, Ladanyi M. Aberrant nuclear immunoreactivity for TFE3 in neoplasms with TFE3 gene fusions: a sensitive and specific immunohistochemical assay. Am J Surg Pathol 2003; 27: 750-761. PubMed
- 9. Hemesath TJ, Steingrimsson E, McGill G, et al. Microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev 1994; 8: 2770-2780. ArticlePubMed
- 10. Argani P, Aulmann S, Illei PB, et al. A distinctive subset of PEComas harbors TFE3 gene fusions. Am J Surg Pathol 2010; 34: 1395-1406. ArticlePubMed
REFERENCES
Fig. 1Representative radiological and histological images of the tumor. (A) Axial view of a three-dimensional sacral magnetic resonance image showing a 5.5×4.7-cm lobulated and expansile mass. (B) The tumor cells exhibit round to polygonal nuclei with eosinophilic cytoplasm and a sheet-like growth pattern. (C) Tumor cells with clear cytoplasm show perivascular arrangements, suggesting a diagnosis of perivascular epithelioid cell tumor. (D) Adjacent bone tissue is infiltrated by tumor cells.
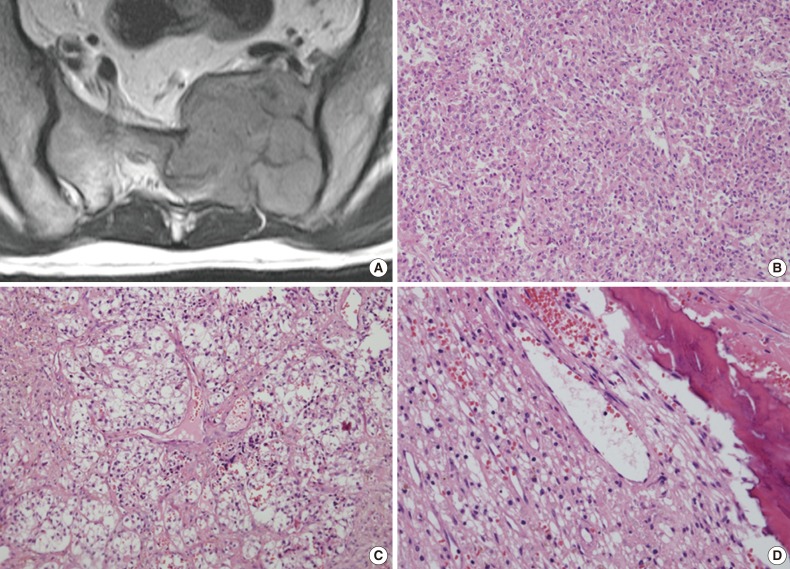
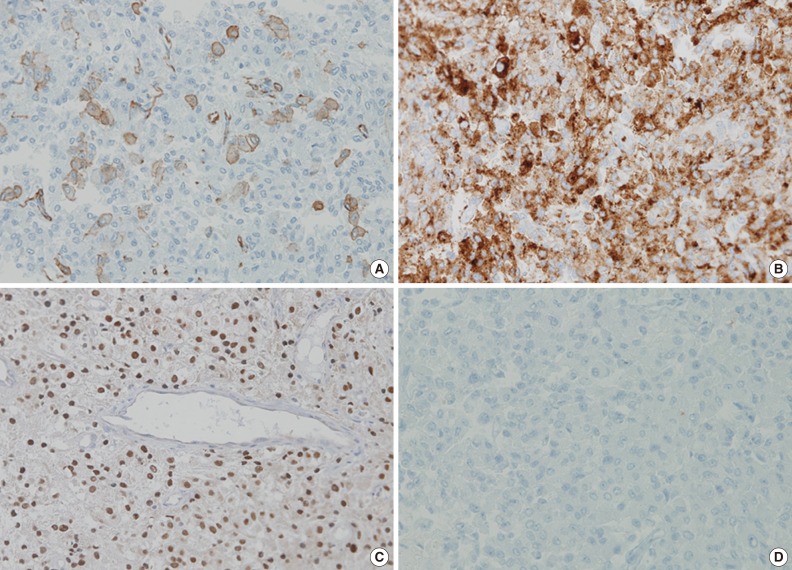
Figure & Data
References
Citations
Citations to this article as recorded by 
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
Perivascular Epithelioid Cell Tumor Arising in the Sacrum: A Case Report


Fig. 1 Representative radiological and histological images of the tumor. (A) Axial view of a three-dimensional sacral magnetic resonance image showing a 5.5×4.7-cm lobulated and expansile mass. (B) The tumor cells exhibit round to polygonal nuclei with eosinophilic cytoplasm and a sheet-like growth pattern. (C) Tumor cells with clear cytoplasm show perivascular arrangements, suggesting a diagnosis of perivascular epithelioid cell tumor. (D) Adjacent bone tissue is infiltrated by tumor cells.
Fig. 2 Results of immunohistochemistry. Smooth muscle actin (A), human melanoma black 45 (B), and transcription factor E3 (C) are expressed in tumor cells. (D) Melan A is negative.
Fig. 1
Fig. 2
Perivascular Epithelioid Cell Tumor Arising in the Sacrum: A Case Report

