 E-submission
E-submission
Search
- Page Path
- HOME > Search
- Single umbilical artery and associated birth defects in perinatal autopsies: prenatal diagnosis and management
- Manushree Saxena, Bhagyashri Hungund
- J Pathol Transl Med. 2024;58(5):214-218. Published online July 9, 2024
- DOI: https://doi.org/10.4132/jptm.2024.07.03

- 25,967 View
- 496 Download
- 2 Web of Science
- 3 Crossref
-
 Abstract
Abstract
 PDF
PDF - Background
The umbilical cord forms the connection between the fetus and the placenta at the feto-maternal interface and normally comprises two umbilical arteries and one umbilical vein. In some cases, only a single umbilical artery (SUA) is present. This study was conducted to evaluate associations between SUA and other congenital malformations discovered in perinatal autopsies and to ascertain the existence of preferential associations between SUA and certain anomalies.
Methods
We evaluated records of all fetuses sent for autopsy to the Department of Pathology during the 10-year period from 2013 through 2022 (n = 1,277). The data were obtained from the hospital’s pathology laboratory records. The congenital anomalies were grouped by organ or system for analysis and included cardiovascular, urinary tract, nervous system, gastrointestinal tract, musculoskeletal, and lung anomalies.
Results
A SUA was present in 8.61% of the autopsies. The gestational age of the affected fetuses ranged between 13 to 40 weeks. An SUA presented as an isolated single anomaly in 44 cases (3.4%). Of the 110 SUA cases, 60% had other congenital anomalies. There was a significant association between birth defects and SUAs (p < .001). Strong associations between SUA and urinary tract, lung, and musculoskeletal anomalies were observed.
Conclusions
A SUA is usually seen in association with other congenital malformations rather than as an isolated defect. Therefore, examination for associated anomalies when an SUA is detected either antenatally or postnatally is imperative. The findings of this study should be helpful in counseling expectant mothers and their families in cases of SUA. -
Citations
Citations to this article as recorded by
- Prevalence of Congenital Abnormalities in Abortuses and Stillbirths in India: A Systematic Review and Meta‐Analysis
Sushant Swaroop Das, Shalika Sharma, Reeha Mahajan, Sushruti Kaushal, Manupriya Sharma, Harsimranjit Singh, M. Ramkumar
Congenital Anomalies.2026;[Epub] CrossRef - Single Umbilical Artery with Symmetrical IUGR and Multiple Fetal Anomalies - An Interesting Case Report
Amulya Choudary Kotapati, Bhargavi Khandru, Vijayasree M.
Journal of Evolution of Medical and Dental Sciences.2025; : 10. CrossRef - Epidemiological and Histopathological Characteristics of Fetuses with Congenital Disorders: A Study in Greece
Despoina Nteli, Maria Nteli, Konstantinos Konstantinidis, Maria Ouzounidou, Paschalis Theotokis, Maria-Eleni Manthou, Iasonas Dermitzakis, Xeni Miliara, Chrysoula Gouta, Stamatia Angelidou, Dimosthenis Miliaras, Soultana Meditskou
Biology.2025; 14(6): 626. CrossRef
- Prevalence of Congenital Abnormalities in Abortuses and Stillbirths in India: A Systematic Review and Meta‐Analysis
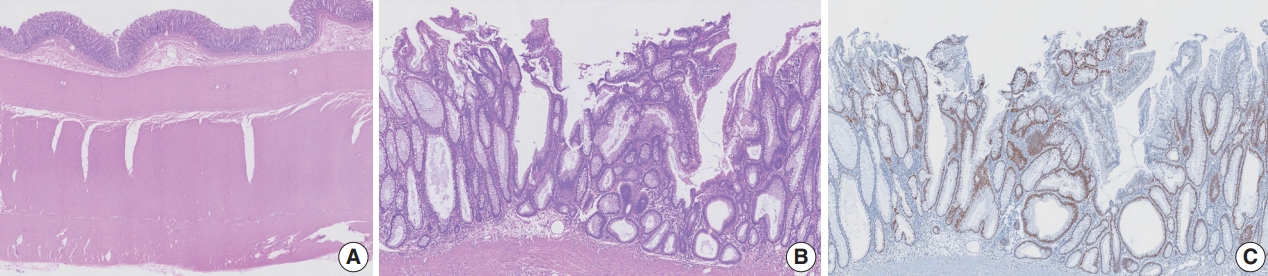
- Tubular adenoma arising in tubular colonic duplication: a case report
- Heonwoo Lee, Hyeong Rok An, Chan Wook Kim, Young Soo Park
- J Pathol Transl Med. 2024;58(4):198-200. Published online July 3, 2024
- DOI: https://doi.org/10.4132/jptm.2024.06.04
- 5,547 View
- 227 Download
- 1 Web of Science
- 1 Crossref
-
Abstract
PDF
- Colonic duplication constitutes a rare congenital anomaly, characterized by the presence of hollow cystic or tubular structures exhibiting an epithelial-lined intestinal wall. Diagnostic challenges persist due to its low incidence and manifestation of nonspecific symptoms such as abdominal pain or constipation, resulting in a reluctance to pursue surgical resection. As associated malignancies in colonic duplication are rare, the inherent malignant potential of these anomalies remains undetermined. Additionally, despite reported instances of associated malignancies in colonic duplication, there is an absence of reports in the literature detailing tubular adenoma within these cases. The histologic features of the presented case are particularly noteworthy, situated at the precancerous stage, intimating potential progression towards adenocarcinoma within colonic duplication.
-
Citations
Citations to this article as recorded by- Low-grade mucinous neoplasm originating from intestinal duplication: a case report and review of the literature
Huihui Yin, Jie Yu, Yunzhao Chen
World Journal of Surgical Oncology.2025;[Epub] CrossRef
- Low-grade mucinous neoplasm originating from intestinal duplication: a case report and review of the literature
- Congenital Peribronchial Myofibroblastic Tumor: A Case Study and Literature Review
- Yuil Kim, Ha Young Park, Junhun Cho, Joungho Han, Eun Yoon Cho
- Korean J Pathol. 2013;47(2):172-176. Published online April 24, 2013
- DOI: https://doi.org/10.4132/KoreanJPathol.2013.47.2.172
- 10,672 View
- 63 Download
- 10 Crossref
-
Abstract
PDF
Congenital peribronchial myofibroblastic tumor (CPMT) is a benign pulmonary spindle cell neoplasm of intrauterine and perinatal period, which is thought to arise from primitive peribronchial mesenchyme. We present a case detected incidentally in a one-month-old infant. The solid and partially necrotic tumor involved the right middle and lower lobes of the lung with extension to the diaphragm. Histologically, the tumor was composed of fasciculated monotonous spindle cells, proliferating peribronchiolar cartilage and round cells with rich vasculature, and high mitotic activity was identified in the round cell area. Immunohistochemical and electron microscopic studies showed that the spindle cells were myofibroblastic in phenotype. Although the tumor showed several malignant pathological features, recurrence was not observed in the two-year follow-up period, consistent with the benign clinical behavior of CPMT.
-
Citations
Citations to this article as recorded by- Congenital Peribronchial Myofibroblastic Tumor: Clinical Features, Pathology, and Surgical Considerations
Kavya Rajesh, Drew Bolster, Mariam Naqvi, Sonya Fabricant, Raghavendra Pillappa, Andrew Brownlee, Carlos Pelayo, Eugene Kim, David Bliss, Eveline Shue
Pediatric Blood & Cancer.2026;[Epub] CrossRef - Congenital Peribronchial Myofibroblastic Tumors Harbor a Recurrent EGFR Kinase Domain Duplication
Sheren Younes, Carlos J. Suarez, Jennifer Pogoriler, Tricia Bhatti, Archana Shenoy, Raya Saab, Lea F. Surrey, Serena Y. Tan
Modern Pathology.2025; 38(2): 100661. CrossRef - EGFR‐KDD Myofibroblastic Neoplasm or Congenital Peribronchial Myofibroblastic Tumor (CPMT)? Report of a Congenital Myofibroblastic Neoplasm With Unusual Histologic Features
Emma Rullo, Sabina Barresi, Sabrina Rossi, Sara Patrizi, Evelina Miele, Marta Barisella, Michela Casanova, Andrea Ferrari, Stefano Chiaravalli, Gloria Pelizzo, Rita Alaggio
Genes, Chromosomes and Cancer.2025;[Epub] CrossRef - Congenital peribronchial myofibroblastic tumor (CPMT): a case report with long term follow-up and next-generation sequencing (NGS)
Ping Zhou, Shuang Li, Weiya Wang, Yuan Tang, Lili Jiang
BMC Pediatrics.2023;[Epub] CrossRef - Neonatal congenital lung tumors — the importance of mid-second-trimester ultrasound as a diagnostic clue
Stephan L. Waelti, Laurent Garel, Dorothée Dal Soglio, Françoise Rypens, Michael Messerli, Josée Dubois
Pediatric Radiology.2017; 47(13): 1766. CrossRef - Congenital peribronchial myofibroblastic tumor: Case report and review of literature
Jolanta Jedrzkiewicz, Eric Scaife, Bo Hong, Sarah South, Mouied Alashari
Journal of Pediatric Surgery Case Reports.2015; 3(4): 154. CrossRef - Perinatal Thoracic Mass Lesions: Pre- and Postnatal Imaging
Evan J. Zucker, Monica Epelman, Beverley Newman
Seminars in Ultrasound, CT and MRI.2015; 36(6): 501. CrossRef - Prenatal imaging and immunohistochemical analysis of congenital peribronchial myofibroblastic tumor
Y.‐A. Tu, W.‐C. Lin, H.‐J. Chen, J.‐C. Shih
Ultrasound in Obstetrics & Gynecology.2015; 46(2): 247. CrossRef - A Congenital Peribronchial Myofibroblastic Tumor Detected in a Premature Infant at 28 Weeks but That Resolved in the Late Stage of Pregnancy
Bo Xia, Gang Yu, Chun Hong, Lei Zhang, Jing Tang, Cuifen Liu
Medicine.2015; 94(42): e1842. CrossRef - Congenital peribronchial myofibroblastic tumor
Yuka Hotokebuchi, Kenichi Kohashi, Satoshi Toyoshima, Naoko Matsumoto, Toshinori Nakashima, Yoshinao Oda
Pathology International.2014; 64(4): 189. CrossRef
- Congenital Peribronchial Myofibroblastic Tumor: Clinical Features, Pathology, and Surgical Considerations
- Pathologic Review of Cystic and Cavitary Lung Diseases
- Na Rae Kim, Joungho Han
- Korean J Pathol. 2012;46(5):407-414. Published online October 25, 2012
- DOI: https://doi.org/10.4132/KoreanJPathol.2012.46.5.407
- 25,597 View
- 368 Download
- 18 Crossref
-
Abstract
PDF
Pulmonary cystic and cavitary lesions caused by diverse etiologies are commonly encountered in chest imaging. The terms "cyst" and "cavity" are used to describe air-filled regions in the center of a nodule or consolidation of the lung. To date, only radiologic aspects of these lesions have been addressed. The morphologies of pulmonary cystic and cavitary lesions exhibit a broad spectrum, ranging from benign to malignant pulmonary diseases of acquired or congenital origin, including variable infectious diseases. In this review, we summarized the differential diagnosis of pathological entities to provide pathologists and radiologists with an overview of the diseases most commonly associated with pulmonary cystic and cavitary lesions in adults and children. The results showed slightly different patterns in the distribution of the diseases in the two groups. The most common causes of cavitary lesions include malignancy and infection in adults, and congenital malformation in children. Therefore, identification of pathologic entities correlating with the radiologic findings, clinical course, and location of the lesion is important in the evaluation of cystic and cavitary lung lesions in order to avoid unnecessary surgical procedures or delayed treatment.
-
Citations
Citations to this article as recorded by- Cavitary pulmonary metastasis from renal pelvic urothelial carcinoma with pathological evidence of bronchiolar obstruction supporting a check-valve mechanism: a case report
Kei Nakano, Tomoki Nakagawa, Haruka Kishi, Shota Fujino, Takashi Ishihara, Masaya Ohara, Kazuhiro Matsuo, Tomoki Higeta, Kie Maita, Hirohito Kobayashi, Masatoshi Yamada, Ryota Masuda
Respiratory Medicine Case Reports.2026; 62: 102431. CrossRef - Managing Cavitary Coccidioidomycosis Expert Opinions for Improving Patient Outcomes
Fariba M. Donovan, George R. Thompson, Janis E. Blair, Royce H. Johnson, Josh Malo, Waseem Albasha, Stephanie G. Worrell, Staci E. Beamer, Kavitha Yaddanapudi, John N. Galgiani, Neil M. Ampel
CHEST.2025; 167(5): 1311. CrossRef - Causes and outcome of pulmonary cysts in pediatric age group among Egyptian patients, case-series study
Erini Fawzy, Mai El-Mahdy, Nevine Elhelaly, Iman Abdulaziz, Muhammad Hegazy, Amr Mustafa
Egyptian Pediatric Association Gazette.2025;[Epub] CrossRef - Weakly-Supervised Segmentation-Based Quantitative Characterization of Pulmonary Cavity Lesions in CT Scans
Wenyu Xing, Yanping Yang, Yanan Zhou, Tao Jiang, Yifang Li, Yuanlin Song, Dongni Hou, Dean TA
IEEE Journal of Translational Engineering in Health and Medicine.2024; 12: 457. CrossRef - Chemotherapy-induced cavitating Wilms' tumor pulmonary metastasis: Active disease or scarring? A case report and literature review
Angelo Zarfati, Cristina Martucci, Alessandro Crocoli, Annalisa Serra, Giorgio Persano, Alessandro Inserra
Frontiers in Pediatrics.2023;[Epub] CrossRef - High-Resolution Computed Tomography of Cystic Lung Disease
Joanna E. Kusmirek, Cristopher A. Meyer
Seminars in Respiratory and Critical Care Medicine.2022; 43(06): 792. CrossRef - Miliary tuberculosis in a paediatric patient with psoriasis
Jacob Kilgore, Jonathon Pelletier, Bradford Becken, Stephen Kenny, Samrat Das, Lisa Parnell
BMJ Case Reports.2021; 14(3): e237580. CrossRef - Atypical pulmonary metastases in children: the spectrum of radiologic findings
Michal Scolnik, Luda Glozman, Ronen Bar-Yoseph, Michal Gur, Yazeed Toukan, Lea Bentur, Anat Ilivitzki
Pediatric Radiology.2021; 51(10): 1907. CrossRef - Radiographic and CT appearance of cavitary pulmonary lesions in a lamb
J Kan, J Bauquier, D Tyrrell, K O'Byrne, AW Stent, B Brosnan
Australian Veterinary Journal.2021; 99(12): 529. CrossRef - Community-acquired Achromobacter xylosoxidans infection presenting as a cavitary lung disease in an immunocompetent patient
Chan Hee Hwang, Woo Jin Kim, Hye Young Jwa, Sung Heon Song
Yeungnam University Journal of Medicine.2020; 37(1): 54. CrossRef - Clinical Research of Pulmonary Langerhans Cell Histiocytosis in Children
Dong Wang, Lei Cui, Zhi-Gang Li, Li Zhang, Hong-Yun Lian, Hong-Hao Ma, Yun-Ze Zhao, Xiao-Xi Zhao, Tian-You Wang, Rui Zhang
Chinese Medical Journal.2018; 131(15): 1793. CrossRef - Benign features of infection‐related tumor‐like lesions of the lung: A retrospective imaging review study
Chun‐Chao Huang, Sho‐Ting Hung, Wei‐Chin Chang, Chin‐Yin Sheu
Journal of Medical Imaging and Radiation Oncology.2017; 61(4): 481. CrossRef - Cavitary lung disease in renal transplant recipients: A single center experience
Gizem Kumru, Serkan Akturk, Siyar Erdogmus, Aysegul Gursoy Coruh, Acar Tuzuner, Sule Sengul, Kenan Keven
Transplantation Reports.2017; 2(4): 19. CrossRef - Solitary lung cavities: CT findings in malignant and non-malignant disease
C.S. Nin, V.V.S. de Souza, G.R.T. Alves, R.H. do Amaral, K.L. Irion, E. Marchiori, B. Hochhegger
Clinical Radiology.2016; 71(11): 1132. CrossRef - Radial endobronchial ultrasound with a guide sheath for diagnosis of peripheral cavitary lung lesions: a retrospective study
Manabu Hayama, Norio Okamoto, Hidekazu Suzuki, Motohiro Tamiya, Takayuki Shiroyama, Ayako Tanaka, Takuji Nishida, Takashi Nishihara, Nobuko Uehara, Naoko Morishita, Kunimitsu Kawahara, Tomonori Hirashima
BMC Pulmonary Medicine.2016;[Epub] CrossRef - An infant with a hyperlucent chest mass: An unexpected diagnosis
Zarmina Ehsan, Jaimie D. Nathan, Carolyn M. Kercsmar
Pediatric Pulmonology.2015; 50(12): E52. CrossRef - The Pseudocavitation Sign of Lung Adenocarcinoma
Tina D. Tailor, Rodney A. Schmidt, Keith D. Eaton, Douglas E. Wood, Sudhakar N. J. Pipavath
Journal of Thoracic Imaging.2015; 30(5): 308. CrossRef - A Case of Pulmonary Artery Sarcoma Presented as Cavitary Pulmonary Lesions
Daniel Min, Ji-Hyun Lee, Hye-Cheol Jeong, Jung-Hyun Kim, Suk-Pyo Shin, Hong-Min Kim, Kyu Hyun Han, Hye Yun Jeong, Eun-Kyung Kim
Tuberculosis and Respiratory Diseases.2014; 76(3): 136. CrossRef
- Cavitary pulmonary metastasis from renal pelvic urothelial carcinoma with pathological evidence of bronchiolar obstruction supporting a check-valve mechanism: a case report
- Congenital Pulmonary Lymphangiectasia, Associated with Total Anomalous Pulmonary Venous Return.
- Seong Wook Hwang, Mee Seon Kim, Po Eun Park, Tae In Park
- Korean J Pathol. 2011;45(6):650-653.
- DOI: https://doi.org/10.4132/KoreanJPathol.2011.45.6.650
- 4,197 View
- 48 Download
-
Abstract
PDF
- Congenital pulmonary lymphangiectasia (CPL) is very rare. It shows diffuse pulmonary lymphatic dilatation without lymphatic proliferation. CPL can occur as a primary disorder or arise secondarily from other diseases such as the obstruction of pulmonary veins or lymphatics. The prognosis of CPL is very poor. Approximately 50% of infants are stillborn and most others usually die within the first day of life. The present case showed diffuse lymphangiectasia in the subpleural, interlobular, and peribronchovascular areas. The flat lining cells were immunohistochemically positive for D2-40 and CD31. CPL is usually diagnosed by clinicoradiological or postmortem examinations. However, our case was diagnosed by an antemortem lung biopsy. We report a case of CPL with total anomalous pulmonary venous return.
- Coexistence of Intrapulmonary Bronchogenic Cyst and Congenital Cystic Adenomatoid Malformation: A Case Report.
- Mee Hye Oh, Eun Ah Jung, Ji Hye Lee, Hyun Deuk Cho, Ki Hyun Seo, Seock Yeol Lee, Young Tong Kim
- Korean J Pathol. 2011;45(1):92-95.
- DOI: https://doi.org/10.4132/KoreanJPathol.2011.45.1.92
- 4,205 View
- 27 Download
- 1 Crossref
-
Abstract
PDF
- Congenital cystic lesions of the lung are uncommon and a conjunction of two or more lesions is very rare. We report here on a case of coexisting intrapulmonary bronchogenic cyst and congenital cystic adenomatoid malformation in a 13-year-old female with a cystic mass in the right upper lobe of the lung. Computed tomography showed a cystic lesion measuring 2.5 cm with an air fluid level and surrounding multicystic lesions in the right upper lobe. On gross examination, the cut surface showed a cystic mass containing inspissated mucinous material, and the cystic mass was surrounded by multiple small cysts. Microscopically, the larger cystic cavity was lined with pseudostratified ciliated columnar epithelium. The submucosal tissue contained mucinous glands and plates of cartilage. The surrounding smaller cysts or irregular spaces were lined with bronchiolar-type respiratory epithelium. We propose that this hybrid lung lesion may represent the missing link in a common embryologic pathway determined by the timing of mesenchymal and epithelial interactions.
-
Citations
Citations to this article as recorded by- Case 2: Coexisting Cystic Lesions of Lung in a Term Neonate: A Management Dilemma
Bichitrananda Raut, Aakriti Soni, Susanta Kumar Badatya, Satish Saluja, Manoj Modi, Arun Soni
NeoReviews.2018; 19(9): e542. CrossRef
- Case 2: Coexisting Cystic Lesions of Lung in a Term Neonate: A Management Dilemma
- Congenital Mesoblastic Nephromas with lmmunohistochemical and Flow Cytometric Analysis.
- Woo Hee Jung, Yee Jeong Kim, Jee Young Han, Woo Ick Yang, Dae Young Kang
- Korean J Pathol. 1995;29(3):303-310.
- 2,114 View
- 16 Download
-
Abstract
PDF
- We reviewed 7 cases of congenital mesoblastic nephroma (4 cases of classical mesoblastic nephroma (CMN) and 3 cases of atypical mesoblastic nephroma (AMN)) using immuno-histochemical and flow cytometric study. Results are as follows. 1) The mean tumor size was 5 (3 to 7cm)cm in CMN and 9 (7 to 10cm)cm in AMN. The AMN revealed hemorrhage and necrosis in two Of three cases. A case of AMN showed cystic change without hemorrhage and necrosis. Mitotic count ranged in 0~4/10HPF in CMN and 20-35/10HPF in AMN. 2) Immunohistochemistry for vimentin was all positive. Actin, desmin were weakly positive in CMN, but negative in AMN. The findings were consistent with myofibroblastic differentiation in CMN and AMN was considered to be the less differentiated form of CMN. 3) Flow cytometiic analysis showed diploidy in two of two CMNs and two of three AMNs. Only one AMN showed aneuploidy with DNA index of 1.41. %SG2M were 8.1 and 15.9 (mean 12.0) in CMN and 16.9, 32.9 and 19.3 (mean 22.9) in AMN, respectively. We concluded that AMN should be distinguished from CMN, clinicopathologically.
- Congenital Cystic Disease of the Kidney overview and a classification.
- Mee Joo, Yeon Mee Kim, Chong Jai Kim, Yeon Lim Suh, Jeong Wook Seo, Je Geun Chi
- Korean J Pathol. 1997;31(3):233-243.
- 2,156 View
- 22 Download
-
Abstract
PDF
- The congenital renal cystic disease encompasses a complex group of pathologic and clinical entities. We retrospectively reviewed 42 cases of congenital renal cystic lesions classified into four Potter types in a series of 2,063 consecutive autopsies from 1981 to 1996. According to our study based on morphologic, clinical, genetic features and associated anomalies, type I and III are relatively compatible with Potter's original definition. However, it was reasonable that type II and IV are classified to the same group because of: 1) very similar histologic findings representing dysplastic kidney, 2) many associated anomalies, 3) no evidence of inheritance, and 4) presence of a combined type. Syndrome associated cysts, such as Meckel-Gruber syndrome, were also separately classified. If the dysplastic evidence was insufficient for diagnosis to the dysplastic kidney in type II and IV, then these cases would be better classified into a cystic disease associated with congenital hydronephrosis. We propose a classification of the congenital cystic disease of the kidney to be: 1) dysplastic kidney, 2) cystic disease associated with congenital hydronephrosis, 3) polycystic kidney, and 4) syndromic cystic disease.
- Congenital Subglottic Stenosis of the Larynx Associated with Tracheoesophageal Fistula: 1 autopsy case.
- In Sook Kim, Tae Jung Kwon, Dong Wha Lee
- Korean J Pathol. 1989;23(3):350-354.
- 2,593 View
- 24 Download
-
Abstract
PDF
- Congenital subglottic stenosis of the larynx is one of the most common cause of chronic airway obstruction im infancy and childhood. It is defined as narrowing of the space bounded inferiorly by the inferior margin of the cricoid cartilage amd superiorly by the insertion of the fibers of the conus elasticus into the true vocal cords. In case we experienced was a female full-term baby delivered by Cesarean section. The stenosis was believed by hypertrophy of stromal soft tissue and cricoid cartilage in the subglottic area. The lesion was associated with tracheoesophageal fistula of H1 type. A brief review of the literature was done.
- Congenital Hepatic Fibrosis with Caroli's Disease.
- Yoon Jung Kim, Soon Ae Oak, In Chul Lee
- Korean J Pathol. 1997;31(3):275-279.
- 2,240 View
- 15 Download
-
Abstract
PDF
- Congenital hepatic fibrosis is an inherited, congenital disorder of the liver, and is occasionally associated with cystic disease of the liver and kidney. We present a case of congenital hepatic fibrosis with Caroli's disease. A 21-year-old woman had suffered from an episodic fever with headaches for 3 years. In laboratory examination, the liver function test was within the normal limits. Esophageal varix was noted by an endoscopic examination. Hepatosplenomegaly and multiple dilated bile ducts were seen by abdominal CT scanning. An orthotopic whole liver transplantation was done. The liver was fibrotic and enlarged. Multiple cystically dilated intrahepatic ducts were noted. Microscopically, diffuse portal fibrosis and widening with proliferation of bile ductules were seen. Intrahepatic bile ducts were markedly dilated and tortuous. The liver cell cords were well preserved.
- Morphologic Difference of the Atrial Chambers and Determination of the Atrial Situs in the Normal and Congenitally Malformed Heart.
- Eo Jin Kim, Jeong Wook Seo
- Korean J Pathol. 2004;38(3):174-180.
- 2,185 View
- 16 Download
-
Abstract
PDF
- BACKGROUND
Identification of atrial situs is the initial step in any segmental analysis and classification of congenital heart malformations. To elucidate the differences for both atria of the normal and congenitally malformed heart, we performed morphological studies on the human heart with or without abnormal laterality syndrome.
METHODS
Five normally formed human hearts, five hearts with right isomerism and five hearts with left isomerism were used in this study. The postero-superior walls of the atrial chambers were examined.
RESULTS
Although the division line of the ventral and dorsal compartments was not as conspicuous as was seen in the right atrium of rat embryo previously studied, this division line existed as a well-developed terminal crest and vestigial structure of the antero-lateral extension of the terminal crest. These structures were noted in the right atrial chambers of normal human hearts and in the bilateral atrial chambers of right isomerism. However, they were totally absent in the bilateral atrial chambers in hearts with left isomerism.
CONCLUSIONS
Our study showed that the right atrial chamber in the normally developed human heart has vestigial components of division between the ventral and dorsal compartment, and hearts with right isomerism and left isomerism have differential development of the ventral and the dorsal compartment.
- Congenital Mesoblastic Nephroma.
- Seok Hoon Jeon, Seung Sam Paik, Nam Hoon Kim, Moon Hyang Park, Jung Dal Lee
- Korean J Pathol. 1997;31(4):375-378.
- 2,247 View
- 14 Download
-
Abstract
PDF
- Mesoblastic nephroma is an important differential diagnosis of a renal mass occurring in the neonatal period or in early childhood. It is a rare monomorphous congenital renal neoplasm most commonly recognized during the first 3 months of life. With the widespread application of ultrasound imaging, many cases are recognized prior to birth. We report a case of mesoblastic nephroma detected by ultrasonograph at 36 weeks of intrauterine fetal life and removed after birth. It showed a well circumscribed, grayish white, solid mass measuring 4x3x2 cm. The tumor was predominantly a classic type with a focal cellular pattern. Immunohistochemical and electron microscopic studies were done.
- Amniotic Band Syndrome: An autopsy case report.
- Hye Seon Ahn, Gil Ro Han, Jin Hee Sohn, Jung Il Suh
- Korean J Pathol. 1989;23(4):482-486.
- 2,154 View
- 18 Download
-
Abstract
PDF
- We report an autopsy case of amniotic band syndrome exhibiting microcephaly, asymmetric encephalocele, microphthalmia, nasal deformity, cleft lip and palate accompanied by left maxillary and zygomatic bone deformities. The amniotic membrane of the placenta was also attached to the herniated brain. The twenty-year-old primigravid mother had no history of taking drug, irradiation, infection or trauma before or during pregnancy.
- Congenital Neuroblastoma of the Adrenal with Metastasis to Liver, Contralateral Adrenal and Pituitary: Report of an autopsy case.
- Na Hye Myong, Sang Yong Song, Je G Chi
- Korean J Pathol. 1993;27(2):169-174.
- 2,559 View
- 22 Download
-
Abstract
PDF
- Neoplasms presenting at birth or within the first month of life are defined as congenital tumors. The principal components of this congenital tumors are neuroblastoma, leukemia, brain tumors and sarcomas. The neuroblastoma is the most common accounting for 15~50% of all tumors in this group. It most often presents with an abdominal mass due to adrenal-retroperitoneal primary or hepatomegaly resulting from extensive metastasis. Most often the primary site is adrenal but other loci include the retroperitoneum, mediastinum, pelvis, etc. This 2-day-old female presented with hepatomegaly and a left adrenal mass at birth, first detected by ultrasonography. On the first day, she suffered from hematemesis and bradycardia. She died on the second day. Postmortem examination revealed massive metastatic tumor nodules in the liver and a well-demarcated round mass, 4 cm, in the left adrenal, with necrosis and hemorrhage. Microscopic findings revealed largely undifferentiated neuroblastoma with focal neuronal differentiation and areas of necrosis and calcification in the background of fine fibrovascular stroma. Other metastatic foci were detected in the right adrenal and pituitary gland.
- Congenital Fiber Type Disproportion Myopathy: A case report .
- Sung Hye Park, Kwang Kuk Kim, Suk Yoon Kang, Shin Kwang Kang
- Korean J Pathol. 1999;33(4):303-306.
- 5,435 View
- 21 Download
-
Abstract
PDF
- Authors report a typical case of congenital fiber type disproportion (CFTD) with unique clinicopathologic characteristics. The patient was a 13-year-old boy who presented with weakness of lower extremities, especially proximal muscle, since his infancy. He has suffered from severe scoliosis which got worse since the age of 12. He showed mild dysarthria, high arched palate, and fish face. All routine laboratory data were within normal limits. EMG findings suggested myopathy. The muscle biopsy revealed fiber type disproportion with type 1 predominance. While most of the type 1 myofibers were atrophic or normal in size, the type 2 fibers showed universal hypertrophy. The difference of mean diameter between the larger and the smaller fibers was 27.9%. The patient's clinicopathologic settings fulfilled the criteria of CFTD.
- Achondrogenesis Type 2: An autopsy case.
- Joon Mee Kim, Young Chae Chu, Soo Kee Min, Hee Jeung Cha, Je Geun Chi
- Korean J Pathol. 1997;31(5):482-488.
- 2,737 View
- 33 Download
-
Abstract
PDF
- Achondrogenesis type 2 is a lethal form of congenital skeletal dysplasia characterized by severe short-limbed dwarfism, decreased vertebral ossification and normal ossification of the skull. We report an autopsy case of achondrogenesis type 2 in a female fetus terminated at 29 weeks of gestation. External morphology revealed a relatively large head, short upper and lower extremities, short neck, and distended abdomen. The x-ray finding showed normal calvarial ossification, hypoplastic ilium and unossified ischium, and metaphyseal flares of the femur and tibia. Histologically, chondrocytes were large and irregular with increased vascularity.
- VATER Association: Three autopsy case reports with imusual defects.
- Mi Ja Lee, Myeong Cherl Kook, Je G Chi
- Korean J Pathol. 1995;29(5):678-683.
- 2,181 View
- 10 Download
-
Abstract
- VATER association represents vertebral defects, anal atresia, tracheo-esophageal fistula with without esophageal atresia, renal defects and radial limb dysplasia. The probability of the simultaneous occurrence of any three of these defects is so unlikely that it suggests a sporadic non-random association. This non-random association appears to be related to some chromosomal anomalies, the caudal regression syndrome, mesodermal defects in early developmental period or the matemal use of sex hormones during embryogenesis. We report three autopsy cases of the VATER association that showed most of the known major and minor defects as well as an unusual concurrence of other defects, i.e., scoliosis, talipes varus, absent penis, urethral agenesis and stenosis, rectourethral fistula, rib anomaly, single umbilical artery, Meckel's diverticulum, diaphragmatic hemia, absent rectum, short neck, simian crease, low set ear, and hypoplastic lung.
- Infantile Myofibromatosis(Congenital Generalized Fibromatosis): Associated with multiple congenital malformations and basaloid follicular hamartomas in the skin.
- Eun Sook Nam, Yoo Hun Kim, Han Kyeom Kim, Insun Kim, Je Geun Chi
- Korean J Pathol. 1995;29(6):776-782.
- 2,446 View
- 19 Download
-
Abstract
PDF
- Infantile myofibromatosis with systemic involvement is a very rare disease and is characterized by numerous nodules composed of spindle cells of a myofibroblastic nature. There are often disseminated throughout the subcutis, muscle, skeleton and viscera. We report an autopsy case of infantile myofibromatosis in a stillborn female fetus of 32 weeks of gestation. The nodules, Imm to 2 cm, were found over the whole body and viscera. The involved viscera were the heart, tongue, esophagus, gastrointestinal tract, portal areas of the liver, spleen anc pancreas. There were also associated malformations, viz., frontal meningoencephalocele, flexion defer-mities, syndactyly, cleft palate, agenesis of corpus callosum, pachygyria, diaphragmatic hemia, renal hypoplasia, etc. Multiple basaloid follicular hamartomas of the skin were noted on the face and extremeties. There are no previous reports in the literature of infantile myofibromatosis in conjunction with the above skin lesion and congenital malformations.
- Congenital Intracranial Teratoma with Extension into Oral Cavity: An autopsy case.
- Young Sill Kim, Kyo Young Lee, Chang Suck Kang, Sang In Shim, Sun Moo Kim
- Korean J Pathol. 1990;24(3):326-330.
- 2,030 View
- 16 Download
-
Abstract
PDF
- Intracranial teratomas which were first described by Maier in 1861 are uncommon. Those presenting at birth are very rare and in our knowledge no case has been reported in Korea. In November, 1988, we experience a case of congenital intracrainal teratoma which replaced almost all cerebral tissue, filled out the oral cavity, and was protruded from the mouth. A female fetus was artificially delivered by a 25-year-old primigravida at 22 weeks of gestation, because of marked hydramnios and fetal hydrocephalus which were detected by prenatal ultrasonography. Microscopically, tissues from all three germ layers, including a lot of neuroepithelim and primitive mesenchymal tissue, were shown. A brief review of the literature was done.
- Laryngeal Atresia with Tracheoesophageal Fistula: 1 case report.
- Eun Kyung Kim, Je G Chi
- Korean J Pathol. 1993;27(5):504-508.
- 2,363 View
- 22 Download
-
Abstract
PDF
- Laryngeal atresia is a very rare congenital anomaly requiring immediate tracheotomy. We present a case of laryngeal atresia with tracheoesophageal fistula who showed immediate respiratory difficulty after ligation of umbilical cord and died of aspiration pneumonia at 8 days of age. The atretic portion of larynx is composed of irregulary arranged cartilaginous tissue, bundles of intrinsic muscle and soft tissue without epithelium-lined lumen. The lungs show normal development and evidences of aspiration pneumonia.
- Myotubular myopathy: A case report.
- Je G Chi
- Korean J Pathol. 1986;20(3):328-331.
- 2,308 View
- 33 Download
-
Abstract
PDF
- A case of a myotubular myopathy in a 5 year old boy is described. This was the first and the only boy to a 30 year old mother who had no prenatal or perinatal problems. No family history of muscle disease was present. His muscle weakness started from neonatal period but was very slowly progressive. The developmental milestones were generally delayed. He had repeated episodes of pneumonia. Muscle biopsy revealed characteristic cental nuclei in 68% of myofibers, and this findings was associated with generally small and round fibers and minimal interstitial change. No inflammatory reaction was present.
- Malignant Melanoma Arising in Giant Congenital Melanocytic Nevus: A case report.
- Jung Sun Kim, Sang Yong Song, Kye Yong Song, Je G Chi
- Korean J Pathol. 1993;27(6):650-655.
- 2,448 View
- 39 Download
-
Abstract
PDF
- Giant congenital melanocytic nevus is found in 0.1% of live born infants. If present, this lesion has a 6.3% chance to develop malignant melanoma. We report such a case in a 22-year-old woman who had multiple pigmented skin lesions since birth. Rapidly growing masses were recently detected in the 19 cm-sized occipital pigmented lesion. Removed scalp lesion revealed yellowish white lobulated soft nodules in the background of pigmented nevus. Microscopically, the nodules consisted of epithelioid cells with prominent nucleoli, and pleomorphic cells including signetring cells. These cells seldom contained melanin pigment. There were metastatic aggregates of tumor cells in the cervical lymph node, which were reminiscent of germinal centers of lymph nodes. S-100 protein immunostaining was helpful to distinguish them. Incidentally, focally scattered pigmented spindle cells were seen in the capsule of a lymph node
- A Sialoblastoma Associated with a Hepatoblastoma: An autopsy case report.
- Sun Lee, Youn Wha Kim, Jae Hoon Park, Yong Koo Park, Ju hie Lee, Moon Ho Yang
- Korean J Pathol. 1997;31(11):1222-1226.
- 2,524 View
- 10 Download
-
Abstract
- Sialoblastoma is defined as a rare, congenital or perinatal, aggressive and potentially low-grade malignant, basaloid gland neoplasm that occurs in the major salivary glands. We report a case of a congenital sialoblastoma in the left parotid gland, associated with a hepatoblastoma in a female infant. At birth, a huge mass in the left neck and hepatomegaly were noted. Grossly, the neck mass was well-circumscribed, lobulated and gray tan. Microscopically, the tumor was composed of basaloid aggregates of primitive uniform cells with focal ductal differentiation. The liver showed a well-circumscribed gray tan tumor with extensive hemorrhage and cystic change. Microscopically, the liver revealed characteristic findings of hepatoblastoma. To the best of our knowledge, this is the first case of coexistence of a congenital sialoblastoma and a hepatoblastoma, reported in the literature.
- Congenital Sialoblastoma: A case report and review.
- Jong In Yook, Hee Jeong Ahn, Jin Kim
- Korean J Pathol. 1997;31(11):1227-1232.
- 2,202 View
- 10 Download
-
Abstract
- A congenital salivary gland tumor, sialoblastoma, is extremely rare. A sialoblastoma of the parotid gland, occurring in a 28-week old fetus, is described. The histologic, immunohistochemical, and ultrastructural features of this tumor were studied. The tumor was characterized by solid nests or sheets of tumor cells intermingled with ductal structures lined by a columnar cells. Some of the tumor cells showed squamous differentiation. Immunohistochemically, these epidermoid cells reacted positively with anti-cytokeratin. But anti-S-100, anti- vimentin, anti-smooth muscle actin, anti-GFAP positive cells were not found. The ultrastructure was characterized by primitive epithelial cells. Although various names have been proposed, we favored the term "sialoblastoma". The histogenesis of this tumor is also discussed.
- Congenital Hepatic Fibrosis: A case report.
- Weon Young Choi, Sun Hee Yoon, In Sook Lim, Ha Jin Choi
- Korean J Pathol. 1991;25(1):50-53.
- 2,218 View
- 13 Download
-
Abstract
PDF
- Congenital hepatic fibrosis is an uncommon disease of children and young adults with two major risks: gastrointestinal hemorrhage caused by portal hypertension, and cholangitis related to bacterial infection of dilated intrahepatic bile ducts. It is characterizeed by stony hard hepatomegaly and portal hypertension with rather well preserved hepatic function and architecture, and frequent association of the renal lesions. We have recently experienced a case of congenital hepatic fibrosis in a 24 year-old Korean male. The chief complaint was hematemesis from esophageal varices. There were marked hepatosplenomegaly, mild pancytopenia and the liver function test was within normal engorgement and dilatation of portal and splenic veins and multiple cysts of both kidneys.
- Cystic Hygroma of the Neck Pathologic study of 26 autopsy cases.
- Yeon Lim Suh, Je Geun Chi
- Korean J Pathol. 1997;31(12):1256-1263.
- 4,539 View
- 137 Download
-
Abstract
PDF
- Cystic hygroma is a congenital malformation of the lymphatic system appearing single or multiloculated fluid-filled cavities, most often around the neck. They often progress to hydrops and cause fetal death, and frequently associated with chromosomal abnormalities and other congenital malformations. The purpose of our study is to delineate the nature of cystic hygroma and determine the relationship between cystic hygroma and associated anomalies including fetal hydrops. We used data from 26 cases of cervical cystic hygroma in autopsy files of SNU Children's Hospital. Most of cystic hygroma were found in stillborn or premature infants. The fetal cases had been dead for a quite a long period since there was discrepancy between the true gestational age and the developmental age estimated from the body length. Of 26 fetuses only 2 were studied chromosomally and both of them showed 45X. Of 26 cystic hygromas 23 occurred in the posterior neck and 3 in the anterior neck. They ranged from 2.5 to 14 cm (mean: 7.9 cm). The cystic hygroma of the posterior neck consisted of two symmetric sacs on both sides and in the nape and extended to the occipital region. The cystic hygromas of the anterior neck were unilateral or bilateral, and multiloculated and extended into the adjacent cheek. Cystic hygromas of posterior neck were always associated with hydrops, while no recognizable hydrops was found in cystic hygromas of anterior neck. The cystic hygromas were larger in patients with severe hydrops than in patients with less severe hydrops. Associated abnormalities, found in 88%, included hydrops fetalis(88%), growth retardation(80%), cardiovascular anomalies(27%), horseshoe kidney(23%), skeletal anomalies(12%) and hypoxic changes(31%) in visceral organs. In summary, when a hygroma is detected during fetal life, careful sonographic examination for associated congenital anomalies, fetal karyotyping and consideration of artificial abortion are indicated.
- Arthrogryppsis Multiplex Congenita: Pathologic examination of three autopsy cases.
- Seung Sook Lee, Je G Chi
- Korean J Pathol. 1994;28(1):56-64.
- 2,149 View
- 18 Download
-
Abstract
PDF
- Three autopsy cases of arthrogryposis multiplex congenita are studied. They were two deadborns and one neonatal death. All of them had characteristic abnormalities involving multiple joints. Neither primary myopathy nor abnormalities of anterior horn cells of the spinal cord were detected in our cases. However, two cases had minor central nervous system anomalies. All four cases showed pulmonary hypoplasia of varying degree. Two of three cases showed facial dysmorphism such as micrognathia and low set ears, and one showed cleft lip and palate. Ventricular septal defect, umbilical hernia and ureteral anomalies were also associated.
- Congenital Choroid Plexus Papilloma: Report of a case.
- Jee Young Han, Jai Hyang Go, Tai Seung Kim
- Korean J Pathol. 1994;28(1):68-72.
- 2,107 View
- 18 Download
-
Abstract
PDF
- The choroid plexus papilloma is a rare tumor. Its incidence is 0.4-0.6% of all intracranial tumors. Most cases of this tumor are found in the young subject, especially infant and childhood but its congenital form is very rare. The clinical and pathologic findings of congenital choroid plexus papilloma are similar to that of any age. But the cilia on the cell surface are common in the former and very rare in the latter. We present a case of congenital choroid plexus papilloma of the lateral ventricle in a 2 month-old male baby. He had suffered from a sudden onset of convulsions and a drowsy mental state for 2 days. The CT scan revealed a large intraventricular tumor in the left lateral ventricle with hydrocephalus. After ventriculo-peritoneal shunt(V-P shunt), his symptoms were improved. But the hydrocephalus was aggravated due to malfunction of V-P shunt, and he recieved the operation after 4 months. The gross examination revealed a large ovoid papillary tumor(4x3x3cm). On light microscopic examination, the tumor showed papillary structure lined by columnar eosinophilic cells. Some cells had cilia. The electron microscopic finding showed intercellular junction, microvilli and cilia. The tumor cells were positive for cytokeratin, vimentin and S-100 protein.
- Adenocarcinoma Arising in Type 1 Congenital Cystic Adenomatoid Malformation: A Case Report and Review of the Literature.
- Jinyoung Yoo, Sun Mi Lee, Ji Han Jung, Myeong Im Ahn, Deog Gon Cho, Seok Jin Kang, Kyo Young Lee
- Korean J Pathol. 2008;42(6):396-400.
- 2,250 View
- 18 Download
-
Abstract
PDF
- Malignancies in congenital cystic adenomatoid malformations (CCAMs) of the lung are rare. We report a 41-year-old male patient with a pulmonary cystic lesion suspicious for CCAM, unrecognized until the patient was 40 years of age, and which subsequently became more consolidated during the interval between initial presentation and surgery. Microscopic examination of the resected specimen revealed features of type 1 CCAM with a mucinous adenocarcinoma, metastatic to the mediastinal lymph nodes. This case illustrates the importance of prompt surgical resection for all suspected CCAMs, especially those discovered in adulthood.
- Sirenomelia: An autopsy case report.
- Yeong Ju Woo, Hye Kyoung Yoon, In Sook Lim
- Korean J Pathol. 1994;28(1):96-98.
- 2,031 View
- 15 Download
-
Abstract
PDF
- Sirenomelia is a severe form of caudal regression syndrome that results in a fusion of the lower extremities, which is not compatible with life. A various spectrum of anomalies affecting primarily the musculoskeletal, genitourinary and gastrointestinal systems also can occur. This rare malformation has a reported incidence rate of approximately 1 in 60,000 births, with a range of 1 to 1 percent of all malformed infants. We experienced a sirenomelic case with combined anomalies of genitourinary, cardiovascular and gastrointestinal systems. Maternal obstetric history revealed oligohydramnios and intrauterine fetal growth retardation, and the baby was spontaneously delivered at 37 weeks of gestational age, but died I hour after birth.
- Congenital Cystic Adenomatoid Malformation of the Lung: Clinicopathologic analysis of 22 cases.
- Young Lyun Oh, Yeon Lim Suh, Je G Chi
- Korean J Pathol. 1994;28(3):219-227.
- 2,301 View
- 10 Download
-
Abstract
- Congenital cystic adenomatoid malformation of the lung(CCAML) is a rare developmental anomaly characterized by an "adenomatoid" hyperplasia of terminal respiratory structures with formation of the cysts of varying sizes. CCAML is separated into three major types based on the gross and microscopic findings. We have analyzed 22 cases of CCAML, those consisted of 6 autopsy cases and 16 surgical specimens. Out of 22 cases, 5 cases were composed of large cysts(type I) and 9 cases had multiple small cysts(type II). Remaining one case revealed features of solid type(type III), and 7 cases were mixed form. There were 16 boys and 6 girls. All cases were below the age of 14 years. There was no clear-cut age difference between different types of CCAML. However, inflammation, fibrosis and pseudostratification of epithelium were often found in older age. All fetal autopsy cases of CCAML had hydrops fetalis and were associated with maternal hydramnios. One case of type III showed definite mucinogenic cells in the cysts unexpectedly, and one case of the mixed form(typeI+II+III) was found in a fetus of 22 weeks of gestational age. Above findings contradicted the classical description of the CCAML, and suggested that arbitrary classification into three types may not be the best way in understanding this condition.
- Congenital Melanocytic Schwannoma in Ankle Joint Potentially Malignant: A case report.
- Jong Tae Park, Chang Soo Park, Sang Woo Juhng, Kyu Hyuk Cho
- Korean J Pathol. 1987;21(4):308-316.
- 2,215 View
- 10 Download
-
Abstract
PDF
- Congenital malignant melanocytic schwannoma in ankle joint was not reported on literature and it was a very interesting case. Light microscopically, melaninladen cells were mixed in abundant wavy spindle cells, some mitotic cells were also observed. Ultrastructurally, melanosomes in variable stages of development were scattered in the cytoplasm which had basal lamina. Collagen bundles were abundant in the intercellular connective tissue. It was histologically malignant tumor and clinically recurred. But in non-congenital potentially malignant melanocytic schwannoma which had been reported, reccurrence or distant metastasis were not noted. So, further clinical survey may be necessary for evaluation of the malignant behavior of this neoplasm.
- Infantile Fibrosarcoma: A case report.
- Chan Pil Park, Geun Shin Lyu, Chan Kum Park, Jung Dal Lee
- Korean J Pathol. 1994;28(3):313-315.
- 2,383 View
- 10 Download
-
Abstract
- Fibrosarcoma in newborns and infants, designated as congenital, infantile, or juvenile fibrosarcoma is an uncommon soft tissue neoplasm occurring most frequently during the first year of life. Infantile fibrosarcoma is associated with favorable clinical behavior that is markedly different from that of adult fibrosarcoma., We report a case of infantile fibrosarcoma occured in a 3-year-old boy presenting as a palpable mass in the left lower extremity since 3 months of life. Histologic findings of the tumor are similar to those of f ibrosarcomas in adult.
- Plexogenic Pulmonary Arteriopathy in Congenital Heart Disease: A Report of Two Cases.
- Seung Yeon Ha, Kook Yang Park, Hyun Yee Cho, Young Ha Oh, Jae Gul Chung, Dong Hae Chung, Chung Yeul Kim, Han Kyeom Kim
- Korean J Pathol. 2002;36(6):412-415.
- 2,502 View
- 24 Download
-
Abstract
PDF
- Hypertensive pulmonary vascular disease can develop in those cases of congenital cardiac shunt in which critical levels of pulmonary artery pressure and flow are reached and exceeded. We have experienced two cases of plexogenic arteriopathy in complex congenital heart disease and tried to evaluate of distribution of arterial lesions by total mapping of the explanted lung. Case 1 and 2 were 12-year-old boy and 36 year-old man. They were treated with combined heart-lung transplantation. Mapping of the both lungs was done, and graded according to Heath and Edward's grading scheme. The elastic pulmonary artery was tortuous, dilated and aortic configuration. Both lungs showed mostly grade 3. Plexiform lesion or veinlike branches of hypertrophied muscular arteries arosed in a lateral branch of a muscular artery that might be proximal to an area of occlusion. Comprising the right and left lung, the right was more severe than the left. By getting closer to the distal part, the grade tended to increase to 4 to 5. By analyzing the pulmonary lobe, severe pulmonary hypertension of grade 4 or 5 was comparatively disseminated throughout the right lung. On the other hand, in the left lung, the grade of the lower lobe was higher than that of the upper lobe, and within the upper lobe, there was a tendency for the grade of inferior segment to be higher than that of the corresponding apical segment.
- Immunoexpressions of Thyroid Transcription Factor-1 and bcl-2 in Congenital Cystic Adenomatoid Malformation.
- Na Rae Kim, Dong Hoon Kim, Gou Young Kim, Dae Shick Kim, Joungho Han
- Korean J Pathol. 2003;37(1):10-14.
- 2,228 View
- 12 Download
-
Abstract
PDF
- BACKGROUND
Congenital cystic adenomatoid malformation (CCAM) is a congenital abnormality of branching morphogenesis of the lung. Thyroid transcription factor-1 (TTF-1) is detected in human respiratory epithelial cells from 11 weeks of gestation, and at full term, TTF-1 expression is confined within type II epithelial cells and in some respiratory nonciliated bronchiolar epithelial cells. Immunoexpression of bcl-2 is intimately related to apoptosis during the development.
METHODS
To elucidate the nature of the lesion, TTF-1 expression was evaluated in twenty-four cases of CCAM (eight cases of type 1 and sixteen cases of type 2) along with immunostaining for bcl-2. For the control group, four cases of fetal lungs (19 week-, 21 week-, 27 week- and 40 week-gestational age) were also evaluated. In all cases of CCAM, TTF-1 was detected in the nuclei of epithelial cells lining the cysts.
RESULTS
TTF-1 was expressed in the majority of the bronchiolar-like epithelial cells of the cysts in CCAM types 1, and 2, where almost 100% of the lining cells of the cysts were TTF-1 positive with variable intensity, while negative TTF-1 expressions were found in the alveolar-like epithelium of the adjacent alveoli or distal nonciliated bronchi. For bcl-2 immunostaining, no lining epithelial cells of the cysts were stained except for the infiltrating lymphocytes. In the control group, strong immunoreactivities found in early fetal stages were absent in the full-term aged lung (40 gestational weeks).
CONCLUSION
These results support the hypothesis that CCAM types 1 and 2 reflect the abnormalities in lung morphogenesis and differentiation that are distinct from those for normally developed alveolar epithelium or adjacent bronchial epithelium, thus retaining the abnormal TTF-1 immunoreactions. Though restricted to CCAM types 1 and 2 in this study, CCAM might be related to TTF-1 rather than apoptosis in the morphogenesis of the developing lung.
- Atypical Mesoblastic Nephroma: Report of a case.
- Jin Man Kim, Dong Wook Kang, Seung Ki Min, Kwang Sun Suh, Dae Young Kang
- Korean J Pathol. 1991;25(6):601-606.
- 2,279 View
- 15 Download
-
Abstract
PDF
- Congenital mesoblastic nephroma(CMN) is an important differential diagnosis of a renal mass occurring in the newborn or in early childhood. It was first described by Bolande as a separate disease entity distinct form Wilms' tumor. In 1974, Beckwith has predicted that this tumor has a pathologic spectrum with classic congenital mesoblastic nephroma at one extreme, unequivocally mallignant spindle cell sarcomas at the other, and intermediate "gray zone" lesions of indeterminate biologic significanse. In 1986, Joshi has described "atypical mesoblastic nephroma" as a potentially aggressive variant of CMN, which shows atypical gross and microscopic features such as hemorrhage, necrosis, high cellularity, and mitotic index. We report of a case of atypical mesoblastic nephroma presenting in a 38 days-old male infant. Grossly, the tumor involved the upper and midportion of the left kidney. On section, the cut surface was fleshy, grayish-white, and homogeneous. Microscopically, the tumorshowed high degree of cellularity and arrangement of fusiform cells in sheets and vague interlacing bundles. The individual tumor cells showed fusiform to oval nuclei, indistinct scanty pale-eosinophilic cytoplasm and many mitotic figures.
- Congenital Cytomegalic Inclusion Disease combined with Hydrocephalus: A case report.
- Kam Rae Cho, Cheol Hee Yun, Sang Pyo Kim, Kwan Kyu Park, Eun Sook Chang, Taek Hoon Kim
- Korean J Pathol. 1994;28(4):439-441.
- 2,131 View
- 18 Download
-
Abstract
PDF
- This is an autopsy-verifed case of the generalized cytomegalic inclusion disease occuring in a male fetus of a weeks gestation. The fetus revealed hydrocephalus and focal necrosis of brain, focal subcapsular necrosis of liver, and the typical cytomegalic inclusion cells having large acidophilic intranuclear inclusions in the liver, brain, kidney, lung, adrenal gland, pancreas and chorionic villi. Prominent extramedullary hematopoiesis was noted in the liver and kidney. Immuohistochemical staining using anti-cytomegalovirus antibody revealed intranuclear or occasionally intracytoplasmic immunoreactivity in brain, liver, pancreas, lung, kidney, and intestine.
- Congenital Esophageal Stenosis due to Tracheobronchial Remnants: A case report.
- Byung Gon Park, Mee Sook Rho, Sang Yong Lee, Seo Hee Rha, Sook Hee Hong
- Korean J Pathol. 1994;28(4):442-444.
- 2,029 View
- 13 Download
-
Abstract
PDF
- Congenital esophageal stenosis due to tracheobronchial remnants is one of main forms of congenital esophageal stenosis, and it was first described by Frey and Duschel in l936. An 18-month-old male presented with underdevelopment and dehydration state due to persistent vomiting several times per day since 3 months after his birth. Esophagogram revealed an elongated and diiated esophagus with marked stenosis at distal portion. Partial distal esophagectomy was performed. Histologically, the thickened esophageal wall is composed of tracheobronchial remnants including hyaline cartilages, mucous glands, and ductal structures lined by ciliated respiratory epithelium under stratified squamous mucosa.
- Agenesis of the Dorsal Pancreas: An autopsy case.
- Won Sang Park, Ki Hwa Yang, Seok Jin Kang, Byoung Kee Kim, Sun Moo Kim
- Korean J Pathol. 1992;26(1):71-75.
- 2,281 View
- 15 Download
-
Abstract
PDF
- Agenesis of the dorsal pancreas is one of the rare congenital anomalies of the pancreas. Six cases of them have been reported. We have experienced an autopsy case of agenesis of the dorsal pancreas associated with fetal death in the uterus. Grossly, the body and tail of the pancreas and uncinate process were not found and those were partially replaced by adipose tissue. No abnormality was noted in the other organs. Microscopically, pancreatic tissue with autolytic change was identified only in the head portion of the pancreas.
- Aqueductal Atresia with Forking Anomaly: Report of 3 cases.
- Na Hye Myong, Mi Kyung Kim, Je G Chi
- Korean J Pathol. 1994;28(5):514-521.
- 2,473 View
- 16 Download
-
Abstract
PDF
- Aqueductal forking was first described by Russell (l949) as a cause of aqueductal obstruction and a form of congenital malformation with simple stenosis, it is a relatively common cause of congenital hydrocephalus not associated with spina bifida or meningomyelocele. Pathologically it is characterized by two distinct channels separated by non-gliotic brain tissue. We describe variable clinicopathologic findings of 3 autopsy cases showing hydrocephaly due to aqueductal atresia with forking case 1 was a 35-week-old female showing Potter's syndrome, dextrocardia, and skeletal anomaly. case 2 was a 29-week-old male abortus with micrognathia, simian crease, club feet, and minor defects of visceral organs. Case 3 was a 32-week-old female abortus with associated anomalies such as a low-set ear, ectopic thymus and thyroid, and Meckel's diverticulum. On serial sections of brain stems of all 3 cases, were seen variably shaped and atretic lumina of aqueducts with distinct two channe1s and intervening brain tissues of normal cellularity.
- Congenital Acute Myelocytic Leukemia: An autopsy case.
- Kyu Rae Kim, Eun Kyoung Han, In Joon Choi, Chang Hyun Yang, Kir Young Kim
- Korean J Pathol. 1988;22(3):308-316.
- 2,374 View
- 12 Download
-
Abstract
PDF
- Leukemia is a rare disease in the newborn infant. We have presented an autopsy case of congenital acute myelocytic leukemia in a female neonate and discussecd with review of literature. At birth, she was relatively in good health with 4.2 kg in body weight except a large cephalhematoma on left parietal scalp and multiple subcutaneous nodules with ecchymosis on entire body surface. Hemoglobin concentration was 12.0 gm/, Hct 34.6% and erythrocyte count was 2.24 millions. Of 212,400 leukocytes/mm2, 47% were myeloblast. Biopsy of skin nodules reveal leukemia cutis, which disappear dramatically with anticancer drug. The infant was expired 12 days after admission due to intracerebral hemorrhage and acute renal failure.
- Congenital Uterine Cyst: A case report.
- Chang Ho Cho
- Korean J Pathol. 1996;30(10):954-956.
- 2,369 View
- 33 Download
-
Abstract
PDF
- I experienced a case of a congenital intramural cyst of the uterine fundus. On examination by light and electron microscope it was composed of a single layer of thin atrophied lining epithelium without secretory activity and was found to be derived from the paramesonephric duct. This case is presented with a brief review of the literature.
- Congenital Epulis: A Report of Two Cases.
- Hye Kun Oh, Chae Hong Suh
- Korean J Pathol. 2003;37(5):355-357.
- 2,134 View
- 14 Download
-
Abstract
PDF
- We present two cases of congenital epulis in female newborns. Congenital epulis is a very rare lesion of uncertain histogenesis. The present lesions were located on the gingiva of the anterior alveolar ridge of the maxilla and the mandible, respectively. Both tumors consisted mainly of large eosinophilic granular cells arranged in solid nests. The neoplastic granular cells showed positive reactions for neuron specific enolase and vimentin in their cytoplasms, while they were entirely negative for other antibodies used in this study.
- Biliary Cystadeoma of the Liver: Report of a congenital case.
- Jai Hyang Go, Young Nyun Park, Woo Hee Jung, Chanil Park
- Korean J Pathol. 1995;29(2):241-243.
- 2,218 View
- 13 Download
-
Abstract
PDF
- Biliary cystadenoma of the liver is a rare multilocular cystic neoplasm of biliary origin. it occurs most often in middle aged women and rarely in children. Histogenesis of this tumor is uncertain. It may be developmental in origin arising from aberrant hamartomatous bile ducts or ectopic rests of embryonic biliary cystadenoma of the liver discovered at 8 month of intrauterine fetal life. This case supports its congenital theory.
- Congenital Anomalies Observed by Autopsies at the Seoul National University Children's Hospital.
- Jin Haeng Chung, Jeong Wook Seo, Chong Jai Kim, Chul Woo Kim, Je G Chi
- Korean J Pathol. 1997;31(2):93-99.
- 2,190 View
- 21 Download
-
Abstract
PDF
- A retrospective analysis was performed on the 968 cases of fetal or pediatric autopsies over five year period (1990-1994), at the Seoul National University Children's Hospital. Age/mode distribution of cases were artificial abortus(30.6%), spontaneous abortus(12.0%), stillbirth(21.9%), neonates(29%), infants(2.8%) and children(0.9%). Male/female ratio was 1.21. Overall incidence of congenital anomalies was 60.8% and 34.0% of all cases had anomalies involving multiple organ systems. Percentage of cases with any anomaly was 71.6% in artificial abortus, 35.3% in spontaneous abortus, 59% in still births, 65.5% in neonates and 38.9% in infant and children. Common organ systems involved were the cardiovascular system (39.0%), musculoskeletal system (23.6%), nervous system (22.6%), gastrointestinal system (19.9%), and urinary system (14.6%). From these results, we found that the congenital anomalies were most significant diseases of the perinatal period and the cardiovascular anomalies were the most common anomalies of them.
 First
First Prev
Prev


