 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 51(3); 2017 > Article
-
Original Article
An Experimental Infarct Targeting the Internal Capsule: Histopathological and Ultrastructural Changes - Chang-Woo Han, Kyung-Hwa Lee, Myung Giun Noh, Jin-Myung Kim, Hyung-Seok Kim1, Hyung-Sun Kim2, Ra Gyung Kim2, Jongwook Cho2, Hyoung-Ihl Kim2, Min-Cheol Lee
-
Journal of Pathology and Translational Medicine 2017;51(3):292-305.
DOI: https://doi.org/10.4132/jptm.2017.02.17
Published online: May 10, 2017
Department of Pathology, Chonnam National University Medical School and Research Institute of Medical Sciences, Gwangju, Korea
1Department of Forensic Medicine, Chonnam National University Medical School and Research Institute of Medical Sciences, Gwangju, Korea
2Department of Medical System Engineering and School of Mechatronics, Gwangju Institute of Science and Technology, Gwangju, Korea
- Corresponding Author Min-Cheol Lee, MD, PhD Department of Pathology, Chonnam National University Medical School and Research Institute of Medical Sciences, 160 Baekseo-ro, Dong-gu, Gwangju 61469, Korea Tel: +82-62-220-4303 Fax: +82-62-220-5697 E-mail: mclee@jnu.ac.kr
© 2017 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
-
Background
- Stroke involving the cerebral white matter (WM) has increased in prevalence, but most experimental studies have focused on ischemic injury of the gray matter. This study was performed to investigate the WM in a unique rat model of photothrombotic infarct targeting the posterior limb of internal capsule (PLIC), focusing on the identification of the most vulnerable structure in WM by ischemic injury, subsequent glial reaction to the injury, and the fundamental histopathologic feature causing different neurologic outcomes.
-
Methods
- Light microscopy with immunohistochemical stains and electron microscopic examinations of the lesion were performed between 3 hours and 21 days post-ischemic injury.
-
Results
- Initial pathological change develops in myelinated axon, concomitantly with reactive change of astrocytes. The first pathology to present is nodular loosening to separate the myelin sheath with axonal wrinkling. Subsequent pathologies include rupture of the myelin sheath with extrusion of axonal organelles, progressive necrosis, oligodendrocyte degeneration and death, and reactive gliosis. Increase of glial fibrillary acidic protein (GFAP) immunoreactivity is an early event in the ischemic lesion. WM pathologies result in motor dysfunction. Motor function recovery after the infarct was correlated to the extent of PLIC injury proper rather than the infarct volume.
-
Conclusions
- Pathologic changes indicate that the cerebral WM, independent of cortical neurons, is highly vulnerable to the effects of focal ischemia, among which myelin sheath is first damaged. Early increase of GFAP immunoreactivity indicates that astrocyte response initially begins with myelinated axonal injury, and supports the biologic role related to WM injury or plasticity. The reaction of astrocytes in the experimental model might be important for the study of pathogenesis and treatment of the WM stroke.
- Experimental animals
- Male Sprague-Dawley rats, 8- to 9-week-old and weighing 200–240 g, were housed two per cage in a controlled husbandry unit with a 12/12 hour light/dark cycle and permitted ad libitum access to food and water. The temperature was maintained at 21 ± 1˚C. A total of 77 rats were used for the experimental study, and they were divided into three groups: 50 rats for light microscopic histological examination, 13 rats for electron microscopic examination, and 14 rats for measurement of infarct volume. The brain was harvested from 3 hours to 21 days after the photothrmobotic ischemia and infarct volumes were measured at 14 days (Table 1). Animal experiments were performed according to the institutional guidelines of Chonnam National University Research Institute of Medical Science and Gwangju Institute of Science and Technology (GIST). All efforts were made to minimize animal suffering and to reduce the number of animals used. Institutional ethical committee approval was obtained prior to the experiments.
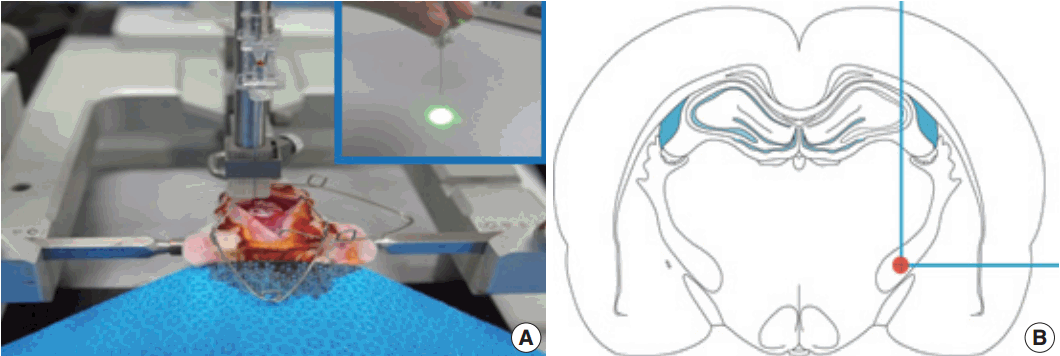
- Induction of IC infarction and behavioral evaluation
- The photothrombotic capsular infarction model used in this study was described previously [21]. Experimental rats were divided into two groups: stroke lesion group (SLG) and sham-operated group (SOG). Briefly, experimental rats of SLG were anesthetized and fixed with a small animal stereotactic frame. A 28-gauge straight stainless steel needle implemented with a fine optical fiber transmitting laser light, a 125 μm outer diameter, and a 62.5 μm core diameter, was inserted through a midline scalp incision. The needle was stereotactically inserted into the PLIC target; 2.0 mm posterior, 3.1 mm lateral to midline, and 7.2 mm deep to bregma, as shown in Fig. 1. The coordinate used in this experiment was deeper than those previously suggested [16]. Rose Bengal dye was injected through the tail vein, followed by 1.5 minutes of laser irradiation with an intensity of 2.5 mW at the fiber tip. After the scalp wound was secured, the rat was transferred to a recovery chamber. The experimental rats of SOG underwent an identical lesion-making procedure except for receiving an injection of saline solution (0.2 mL/100 g) instead of Rose Bengal dye.
- The behavioral tests employed were described in the previous study [21], and single pellet reaching tasks (SPRTs) were used to measure motor performance in this study. Briefly, an experimental apparatus was made of clear Plexiglas with a 1-cm wide slit in the middle of the front wall for the food shelf. Once rats began to show a forelimb preference to obtain a sucrose pellet (Bio-Serve, Frenchtown, NJ, USA) through the slit, a pellet was obliquely placed contralateral to the preferred paw to prevent the use of the nonpreferred paw. Animals were subsequently administered 20 pellets per session for 21 days. Successful reach was defined as a reach in which an animal grasped a food pellet, brought it into the cage, and consumed it without dropping. Reaching performance was scored and the success rate was calculated as the formula, number of successful reaches × 100/20 scores before the operation were averaged to represent preoperative reaching performance. After the photothrombotic infarct, the experimental rats were subdivided as follows from the SPRT score: early recovery group (ERG) characterized by > 50% recovery in 7 days; moderate recovery group (MRG) characterized by > 50% recovery of the prelesion status in 14 days; and poor recovery group (PRG) which had reaching performance at < 50% of the prelesion status. Subjects in the ERG were excluded from the study. Persistent motor impairment, characteristic of both MRG and PRG subjects, developed effectively in 71% of rats. SOG rats did not show a significant decrease in SPRT performance within the observation period.
- Measurement of infarct size
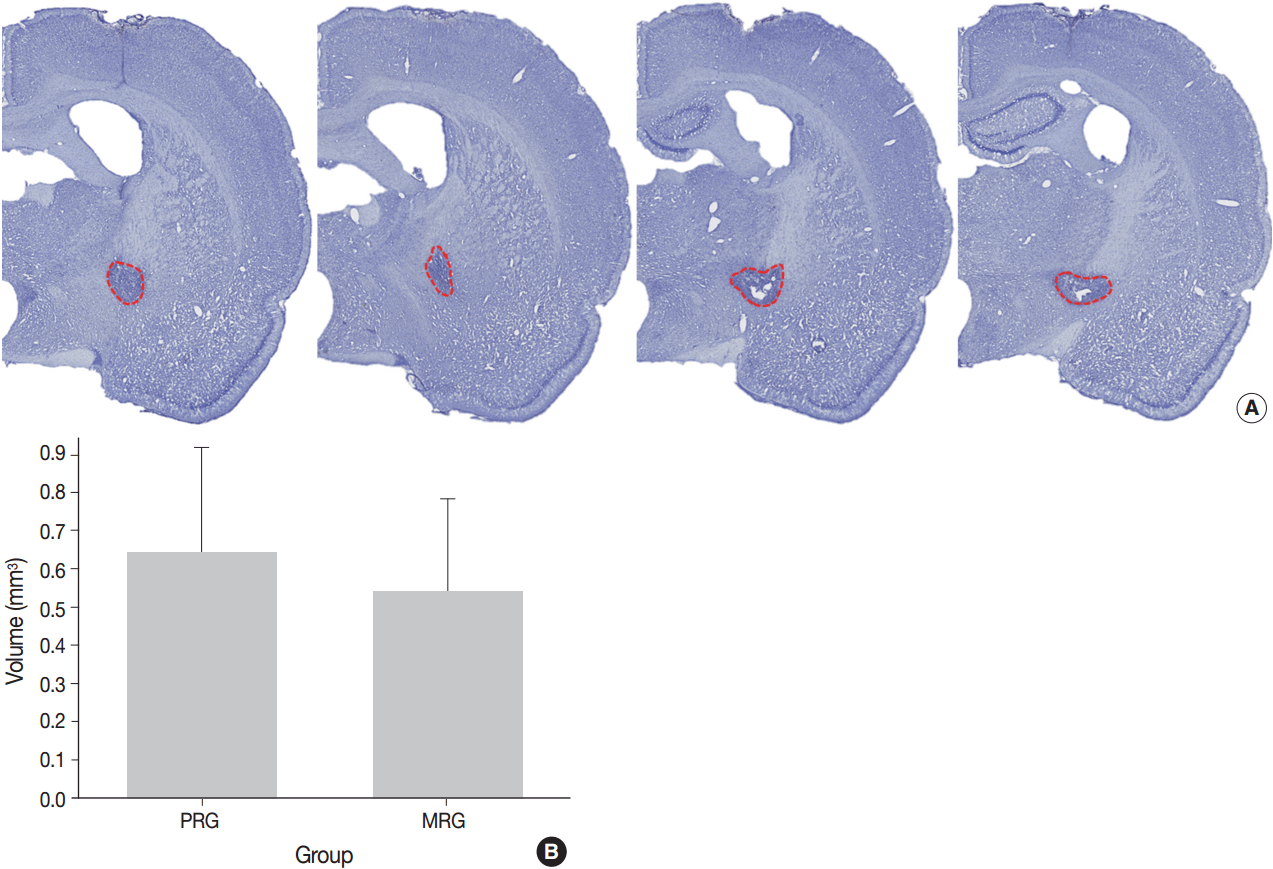
- To measure the infarct size in both MRG and PRG rats, seven fresh brains were perfused at 14 days after the ischemia. Coronal brain sections of 2.5-mm thickness were obtained from the center of the needle insertion sites using a metallic tissue plate covered with optimal cutting temperature (OCT) compound and immediately frozen at –25˚C in a frozen tissue microtome. Serial tissue slices from the frozen brains (20-µm thick sections at 200-µm intervals) were prepared and stained with hematoxylin and eosin (H&E) and cresyl violet. Histologic images were scanned with a ScanScope virtual microscope (Aperio Technologies, Vista, CA, USA). Images from five to six sections were assessed for infarct lesions with Image J software (NIH, Bethesda, MD, USA). The total volume of brain injury was determined by summing the area of damaged tissue recorded from all sections and multiplying by the distance between sections [23]. The completeness of the IC destruction was determined by three of the authors, all of whom were blinded to the motor recovery group.
- Histopathology and immunohistochemistry
- Experimental time-points after PLIC ischemia were as follows. To assess early changes of PLIC infarct, n = 2 each at 3, 6, and 12 hours, and n = 4 each at 1, 4 and 7 days. To assess late changes between MRG and PRG infarcts, n = 8 each at 14 and 21 days. For SOG, n = 2 for each postlesion time-points. After the behavioral evaluation, rats were anesthetized with intraperitoneal sodium pentobarbital (70 mg/kg) and killed by intra-aortic perfusion of 1% paraformaldehyde in phosphate buffer solution (PBS; 0.12 M, pH 7.4) delivered by a perfusion pump (36 mL/min) for 5 minutes followed by 4% paraformaldehyde in PBS for 15 minutes at the same flow rate. The brains were then removed and coronally sectioned into 4-mm-thick slices surrounding the center of the needle insertion site. For light microscopy examinations, the brains were removed from the skull, postfixed in 4% paraformaldehyde for 2 days at room temperature, and subsequently kept in processed paraffin-embedded tissue blocks. Routine H&E, luxol fast blue–periodic acid-Schiff (LBPAS), and immunohistochemical stainings were performed on tissue sections (6-µm-thick) on probed-glass slides. Immunohistochemical staining with anti–glial fibrillary acidic protein (GFAP; 1:400, polyclonal, Zymed, South San Francisco, CA, USA) and anti–neurofilament protein (NFP; 1:600, clone 2F11, Dako Cytomation, Carpinteria, CA, USA) was done using the avidinbiotin peroxidase method [24] with the ABC kit (Vector Laboratories, Burlingame, CA, USA). Antigen retrieval was conducted by heating the tissue sections at 60˚C for 5 minutes in sodium citrate buffer, after which the sections were incubated in a mixture of 3% hydrogen peroxide and 10% methanol for 20 minutes, followed by 10% goat serum for 1 hour at room temperature. The sections were incubated with each primary antibody for 2 hours, biotinylated secondary antibody for 20 minutes, and streptavidin-horseradish peroxidase (Dako Cytomation) for 20 minutes. They were then developed with a chromogenic solution of diaminobenzidine and counterstained with hematoxylin to visualize cell nuclei. Known positive and negative tissues were used as controls.
- Electron microscopy
- One rat each at 3, 6, and 12 hours, and two rats each at 1, 4, 7, 14, and 21 days after the ischemia (n = 13) and one SOG rat were studied by transthoracic cardiac perfusion [25]. Rats were perfused with 1% paraformaldehyde/0.1% glutaraldehyde in cold phosphate buffer (PB) delivered by a perfusion pump (36 mL/min) for 5 minutes, followed by 2% paraformaldehyde/2% glutaraldehyde in cold PB at the same flow rate for 5 minutes. Brains were removed and coronally cut into 2-mm-thick slices. After the lesion was evaluated macroscopically in the brain slices, tissues were trimmed to approximately 2 × 2 × 1 mm, placed in 4% glutaraldehyde in PB for 24 hours at 4˚C, postfixed in 2% osmium tetroxide for 2 hours at room temperature, rinsed in 0.1 mol/L cacodylate buffer, dehydrated in graded concentrations of ethanol followed by propylene oxide, incubated in a mixture of epon/propylene oxide (50/50, then 80/20 for 4 hours), and embedded in epon at 60˚C in an oven for 2 days. Semi-thin sections (1-μm-thick) were stained with 1% toluidine blue and screened by light microscopy to select thin section areas from multiple blocks at each lesion time-point for electron microscopy. Selected thin sections (800-Å thick) were cut with a diamond knife on an ultramicrotome (LKB-Produkter, Bromma, Sweden), mounted on copper grids, contrasted with uranyl acetate and lead citrate, and viewed on a JEOL 120S electron microscope (Nihon Denshi, Tokyo, Japan). The investigators were initially blinded to the treatment conditions. Axon and myelin thicknesses were quantified from 3 hours to 21 days after ischemia. Two random fields per grid from two grids were analyzed per experimental brain. Four random images within each field were collected at × 5,000 magnification and analyzed with ImagePro Plus (MediaCybernetics, Rockville, MD, USA). The axon and myelin sheath diameters were determined for every myelinated axon from a minimum of 30 axons for each experimental group. The myelin thickness was calculated by subtracting the axon diameter from the total fiber diameter. The g-ratio of each myelinated axon was calculated by dividing the average axon diameter by the average total fiber diameter [26].
- Statistical analysis
- Data were analyzed with SPSS ver. 10.0 statistical analysis software (SPSS Inc., Chicago, IL, USA). SPRTs for skilled reaching performances were analyzed with a repeated measure analysis of variances for group and time. Electron microscopy data (g-ratio, axon caliber, and myelin thickness) were tested with a one-way ANOVA. Data are presented as the mean ± standard error of mean, with p < .05 considered significant.
MATERIALS AND METHODS
- Infarct volumes
- A series of frozen tissue slices stained with cresyl violet to measure infarct volume is illustrated in Fig. 2A. Infarct volume included the area of necrosis and surrounding demyelination and astrogliosis. At 14 days after ischemia, the infarct volume was 0.6 ± 0.2 mm3 in the PRG and 0.5 ± 0.2 mm3 in the MRG (Fig. 2B). Infarcts in the PRG were slightly larger than those in the MRG; however, there was no significant relationship between infarct volume and the degree of motor recovery in each group.
- Light microscopy findings
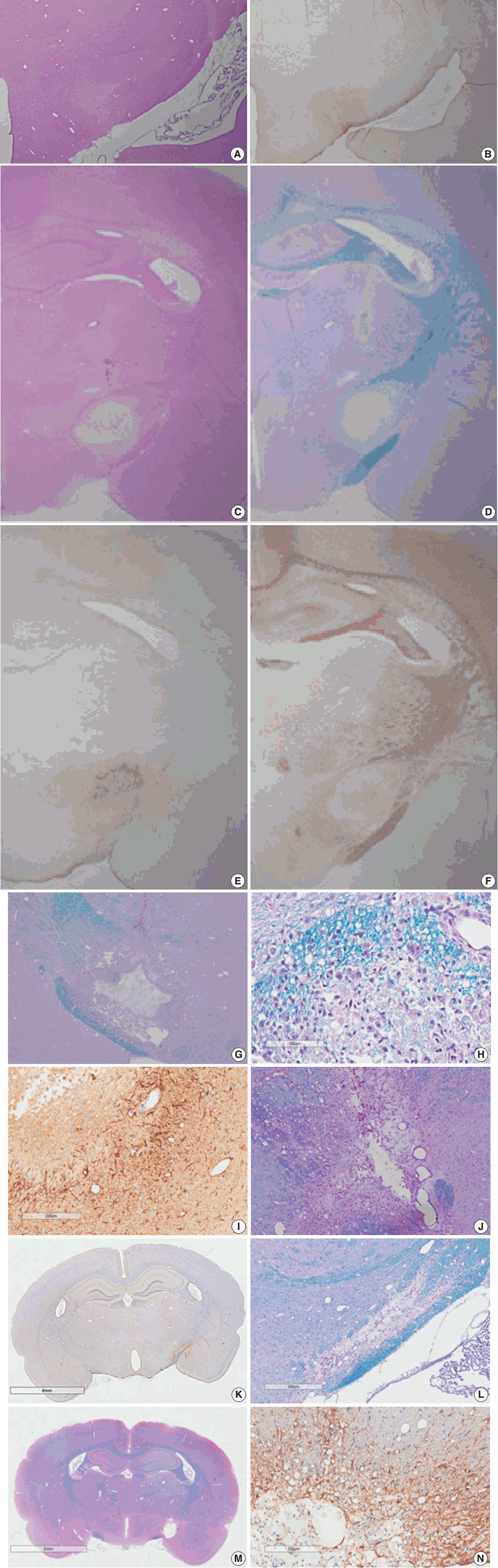
- Myelinated axons in the PLIC were well defined by LBPAS and NFP immunohistochemical staining. The presence of reactive astrocytes was clearly indicated by GFAP immunohistochemical staining. At 3 hours post-ischemia, a pale focal lesion was observed on H&E stained preparation (Fig. 3A). The lesion boundary was not clear and gradually merged with the neighboring normal WM. The lesion was highlighted by light brown positivity on GFAP staining (Fig. 3B). Focal lesions of myelin pallor showed separated, swollen myelinated axons in edematous PLIC by LBPAS staining. Nuclear pyknosis of oligodendrocytes was identified in the lesion. Edematous change was noted in the upper portion of the optic tract and was limited to the vicinity of PLIC.
- At 6 hours to 1 day poststroke, the focal lesion of PLIC was more clearly identifiable with time through low magnification microscopy. A focal infarct manifested as a well-demarcated, pale, whitish lesion due to the lack of staining with H&E and LBPAS (Fig. 3C, D). Immunohistochemical staining revealed increased positivity for GFAP (Fig. 3E) and decreased positivity for NFP (Fig. 3F). Immunopositivity for GFAP in the infarct lesion indicated the reactive change of astrocytes. No inflammatory cell infiltration in the infarct lesion was noted.
- At 4 days post-ischemia, myelinated axons in the PLIC were completely lost by LBPAS staining (Fig. 3G). The lesion had a central geographic necrosis surrounded by a wall of macrophages, in which each macrophage had a central nucleus with prominent nucleoli and abundant cytoplasm phagocytized with light-blue or reddish granular debris (Fig. 3H). Loss of NFP staining in the PLIC also indicated lesion developed. Neutrophil did not infiltrate in the lesion. Strong GFAP staining highlighted presence of hypertrophic astrocytes along the periphery of the infarct lesion and neighboring WM (Fig. 3I).
- At 7 days post-ischemia, the necrotic center showed a decreased volume, and was surrounded by infiltrating macrophages, reactive hypertrophic astrocytes, and newly formed capillaries. Infiltrating macrophages actively phagocytized reddish granular debris throughout the infarct lesion, clearly shown by LBPAS staining (Fig. 3J). At 14 to 21 days post-ischemia, central necrosis was distinctly resolved and the lesion was a pseudocystic with loosely arranged cellular elements. The lesion consisted of a mixture of foamy macrophages, hypertrophic astrocytes with elongated processes, and newly formed capillaries. Critical histopathological features related to motor dysfunction differences between the MRG and PRG were investigated. A portion of myelinated axon bundles passing the PLIC was preserved, as illustrated by immunohistochemical stains for GFAP and NFP and LBPAS staining (Fig. 3K, L). However, the fiber tract of myelinated axons forming the PLIC was severely disrupted and formed pseudocysts in the PRG group, which was clearly identified by LBPAS staining at low magnification (Fig. 3M). In the interpretation of PLIC injury by light microscopy, careful examination of serial tissue sections is important. At 21 days post-ischemia, the focal infarct lesion persisted as a pseudocystic cavity. A few macrophages were still observed along the inner side of the angiogliotic cavity (Fig. 3N). Microscopic examinations confirmed the presence of PLIC lesions (Table 2). The optic tract adjacent to the IC was partially injured in a few rats. However, the adjacent striatum and anterior limb of the IC were intact. The SOG did not show any damage within the underlying IC.
- Ultrastructural findings
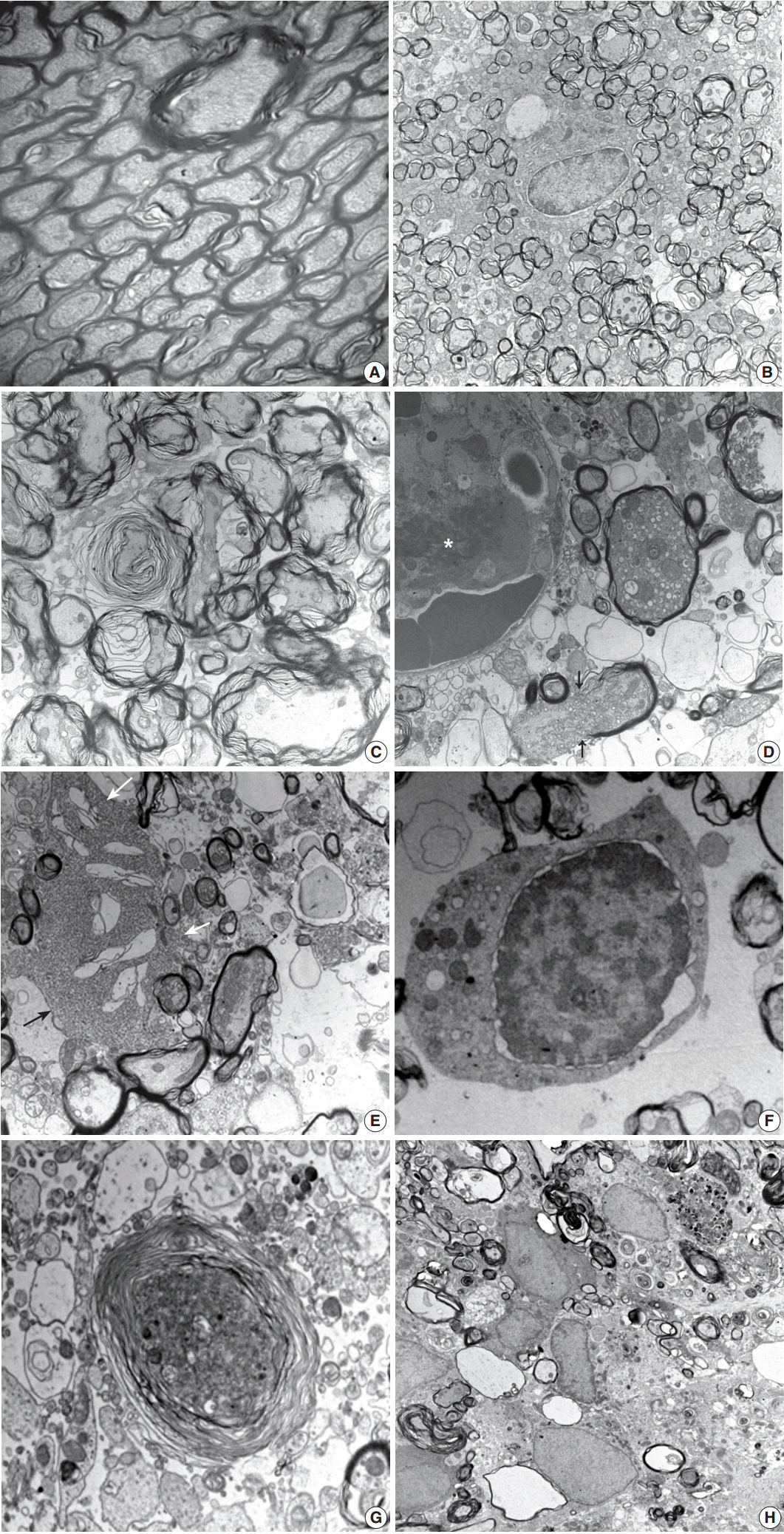
- Histopathological alterations resulting from early ischemic injury were further studied by electron microscopy (Table 2). In SOG, normal myelinated axons presented as a unit of a compact lamellar sheath closely encompassing an axon, and oligodendrocytes were surrounded by myelinated axons (Fig. 4A). Size measurement revealed two types of myelinated axons: large (0.85 ± 0.18 µm diameter) and small (0.41 ± 0.13 µm). Even large axons presented in groups or singular aligned intimately with small axons. Each axon was filled with organized intermediate filament structures; called neurofilaments, and mitochondria were occasionally identified within axons. Progressive structural abnormalities in myelinated axons were observed within the ischemic lesion.
- At 3 hours post-ischemia, platelets aggregated in the capillary lumen and exudative changes were present in interstitial spaces among myelinated axons. Nodular swelling with loosening of the myelin sheath and irregular wrinkling of axons was observed in both large and small myelinated fibers (Fig. 4B). Nodular loosening of the myelin sheath led to the formation of lamellar knob. With nodular loosening of myelin, a clear space developed in the submyelin area and contacted wrinkled axons with polygonal outlines. However, axonal integrity consisting of filamentous structures and mitochondria was relatively well preserved. Oligodendrocytes had relatively intact nuclei, rough endoplasmic reticulum (ER), and mitochondria. Myelin pallor observed by light microscopy in the early stage of the ischemic injury matched the ultrastructural edematous change in the myelin sheath, submyelin space, and wrinkled axons.
- At 6 hours post-ischemia, whorled myelinated axons were formed by progressive loosening of the compact myelin lamellae covered with wrinkled axons (Fig. 4C). Large and small axons identified in SOG rats did not distinct significantly in MRG and PRG groups due to irregular loosening of the myelin sheath and markedly swollen axons. Small hydropic vacuoles developed, and intermediate filaments dissolved in wrinkled axons. At 12 hours to 1 day post-ischemia, the capillary lumen was obliterated by fibrin thrombosis. Nodular loosening of the myelin sheath decreased markedly, and wrinkled axons changed to plump, swollen axons. Swollen axons were filled with many mitochondria, vesicles, and granules, and showed mildly increased electron density (Fig. 4D). With rupture of the myelin sheath,
- axonal organelles (mitochondria, neurofilaments, and vesicles) extruded. Large vacuoles formed by demyelination with axonal flooding were common. Expansion of the extracellular space containing edema fluid presented. Reactive astrocytes were identified by clubbed hypertrophic processes containing abundant intermediate filaments that intimately connected with swollen axons (Fig. 4E). Oligodendrocytes had variable pathologic features ranging from intact nuclei and mildly swollen ER in the cytoplasm to peripheral clumping of coarse chromatin along the nuclear membrane. Oligodendrocytes undergoing apoptosis in the ischemic lesion were identified by irregular clumping of coarse nuclear chromatin at the periphery of the nucleus, detachment of the nuclear membrane, and an electrondense cytoplasm filled with swollen mitochondria and ER (Fig. 4F).
- At 4 days post-ischemia, whorled myelinated axons were present though the pattern differed from those at 6 hours. The whorls were formed by swollen axons filled with cell organelles and electrondense fine filamentous materials covered with whorled lamellae of myelin sheaths (Fig. 4G). Demyelinated ghost axons packed with edematous flooding were common. Macrophages had phagocytized degenerated and demyelinated axons and electrondense bodies (Fig. 4H).
- At 7 to 21 days post-ischemia, the exudative reaction subsided progressively. A number of unmyelinated or thinly myelinated axons were evident. Variably degenerated myelinated axons with an infiltration of scavenging macrophages were seen in the presence of hypertrophic astrocytes and oligodendrocytes. The ultrastructural features were compatible with a pseudocystic wall observed by light microscopy. The number of macrophages decreased with time, but hypertrophic astrocytes were still evident at 21 days post-ischemia.
- Axon and myelin thickness was further separately assessed to identify their contributions to g-ratio changes. Axon diameter significantly decreased (p < .001) at 3 hours to 6 hours post-ischemia. However, myelin thickness (calculated by nerve fiber diameter minus axon diameter) remarkably increased (p < .001) at 3 to 6 hours, and then decreased at 24 hours post-ischemia. The increased axon diameter and decreased myelin thickness at 4 to 21 days post-ischemia contributed to an increased g-ratio in the experimental animals. G-ratios did not differ between sham subjects and the contralateral PLIC in the experimental group.
RESULTS
- In this experiment, a focal lesion in the PLIC was successfully created by laser light with a low intensity light source (814 mW/mm2) and a light delivery needle (28-gauge). The light scattering capacity is 44 times higher in the GM than in WM [27], which might be a promising mechanism to induce a WM infarct in this study. This system created a focal infarct in the PLIC with a chronic loss of motor function, and minimized injury to the adjacent striatum and optic tract. Thus, unwanted neurological and behavioral manifestations did not occur during the experimental period. It was possible to study progressive histopathological changes in the ischemic infarct by light and electron microscopy.
- The human cerebrum is composed of 40% GM and 60% WM, and WM damage causes clinically significant neurological diseases including stroke and vascular dementia [12]. The main components of the WM; myelinated axons, astrocytes, and oligodendrocytes, were injured by photothrombotic ischemia and infarction. In this study, pathological features developed during the first 3-hour highlighted nodular loosening of the myelin sheaths with polygonal wrinkling or compression of the axons. The lesion might be caused by post-thrombotic edema of the WM, indicating that the myelin sheath is very vulnerable to ischemic injury. Concurrent decreases in axonal diameter and increases in myelin sheath thickness at 3 to 6 hours post-ischemia indicated that injury of the myelin sheath and axon were synchronized.
- During the acute phase of ischemic injury, motor dysfunction developed through the damage to myelinated axons, observed by electron microscopy at first. Simultaneously, immediate reactive change of astrocytes, evidenced by increased GFAP immunopositivity in the lesion center, began to recover the damaged WM. Reactive astrocytes continuously presented at 21 days after the WM ischemia in this study. Traditionally, reactive astrogliosis were thought to be detrimental because they secreted inhibitory substrates and provided adverse mechanical barrier for the axonal and dendritic plasticity [28]. However, there were recent studies that reactive astrocytes might provide endogenous mechanisms to protect the ischemic injury and facilitate neural regeneration by releasing many trophic factors [29,30]. Experimental data of our on-going study, data not shown, proved by quantitative morphometric analysis that reactive astrocytes produce highly increased immunoreactivity for brain-derived neurotrophic factor (BDNF) and develop other cerebral lesion by neurotransmitter modification without direct focal ischemic injury, so called diaschisis [31]. BDNF-immunopositive reactive astrocytes facilitated to restore myelin integrity [32] and promoted oligodendroglial differentiation from oligodendroglial precursor cells (OPCs) after WM damage [33]. Intravenous injection of synthetic BDNF also assisted OPC maturation, oligodendrocyte replacement and WM remyelination in WM stroke model of rats [34].
- With the progression of injury to myelinated axons and oligodendrocytes, a focal cystic infarct lesion developed. Cystic infarct in WM were formed by swelling and rupture of myelinated axons, expansion of the extracellular space containing edema fluid, and subsequent loss of massively swollen cellular elements. Macrophages infiltrated the lesion at 4 days post-ischemic injury and phagocytized axon-myelin debris and fat globules. A number of cystic ghost axons also indicated simultaneous injury of axon and myelin sheath by ischemia. Infarct volumes did not differ between subjects in the MRG and PRG. Motor recovery in the two groups depended on the completeness of histopathological capsular injury induced by the ischemia. Subjects in the PRG had more complete thickness destruction of the fiber tract at the anterior PLIC, whereas those in the MRG had partial destruction of the IC in serial sections of the brain. The procedure was clinically well tolerated throughout the experimental period, which spanned from 3 hours to 21 days post-ischemia. These findings show that photothrombotic focal cerebral ischemia produce a highly reliable model of WM infarct in the PLIC.
- The pathophysiological mechanisms of WM stroke were previously most comprehensively studied by ET-1 injection into rat brains [15-19]. The histopathological features differed according to the first component (axon or myelin) injured and experimental methods used. ET-1 impeded regional cerebral blood flow without disintegrating the blood-brain barrier and led to myelin disruption. Axonal injury was evident prior to any clear changes in myelin in the rat model of ET-1 induced WM ischemia, which is a hallmark of WM stroke [15]. Sozmen et al. [35] reported signs of early axonal damage that were observed through ultrastructural electron microscopy studies at 24 hours post-ischemia, whereas myelin sheaths appeared intact within the infarct. Strokes induced by ET-1 injection into the IC are characterized by a well-demarcated, focal, demyelinated lesion and necrosis after 14 days [16]. In this study involving photothrombotic ischemia, concomitant axonal and myelin damage were observed by light and electron microscopy. Although ultrastructural features of whorled myelin formation and polygonal wrinkling of axons appeared simultaneously in this study, edematous change of the myelin sheath was the first pathologic feature by WM ischemia. The photothrombotic model of WM ischemia was not comparable to the ET-1 model, but the histopathological features are significantly similars to those of human WM stroke.
- The inflammatory response following photothrombotic focal ischemia in the GM and WM is contested. A previous study reported prominent neutrophilic infiltrations in the GM at 24 to 48 hours post-ischemia, and cuffing of macrophages surrounding the central necrosis at 4 days post-ischemia [15,16]. However, prominent macrophage infiltration without neutrophil infiltrations was present at 4 days post-ischemia. Even when the same amount of ET-1 was administered to rats, a prominent macrophage/microglia response without neutrophil recruitment in the striatum at 3 days [15] and a proliferative inflammatory response of both neutrophil and mononuclear leukocytes at 1 day post–ET-1 injection have been reported [18]. The difference in inflammatory cell types might be related to cytokines released from the damaged WM and GM and should be studied further. Astrocyte activation occurred early and appeared consecutively with myelin and axonal injury after photothrombotic ischemia. However, increased GFAP staining for astrocyte activation occurred later (7 to 14 days) than the early axonal damage in ET-1–induced subcortical/WM stroke models [15]. In this experiment, increased GFAP staining indicated that the astrocyte response was limited to the PLIC lesion and appeared very early at 1 hour post-ischemia. The number of astrocytes did not change, and cell hypertrophy increased GFAP staining in the lesion. GFAP staining delineated the lesion site persistently during the experimental period. The GFAP-positive astrocyte response was similar to that in the photothrombotic and ET-1 induced WM strokes in rats. Increased the number of astrocytes with GFAP positivity also observed in the ET-1 mouse model [35].
- The AchA is a branch of the internal carotid artery [14], and intraluminal occlusion of the AchA in rats resulted in variable infarct size in the IC and hypothalamus [36]. A study reported the comprehensive histopathology of IC lacunar infarcts by AchA occlusion in gyrencephalic brains of miniature pigs. The study defined the area of the ischemic lesion as the ischemic core (an irreversibly damaged area), peri-infarct area, and marginal zone [26]. Light and electron microscopy of the AchA occlusion model showed progressive histopathology of edema, disrupted axonal alignment, and aberrant myelin sheaths at 24 hours post-ischemia. GFAP-positive astrocytes in the peri-infarct area from early stages, infiltrated macrophages that phagocytized myelin debris and swollen axons, and several organelles within fragile myelin sheaths were also observed at 1 week post-ischemia. The most important finding from the ultrastructural analysis was that axonal damage resulted in myelin sheath instability and subsequent demyelination. These findings corresponded with the early and late histopathological changes found in the photothrombotic capsular infarct model used in this study. Ischemia disturbs ion homeostasis due to a loss of ATP, which results in Ca2+ overload in axons and glia. This process is accelerated by glutamate-mediated over-activation of ionotropic receptors. An initial pathology of axon and myelin swelling in WM ischemia is caused by excess glutamatergic activation of the α-amino-3-hydroxy-5-methyl-4-isoxzole propionic acid (AMPA) receptors in the axon, while excitotoxic activation of N-methyl-D-aspartate receptors is the initial event in GM ischemia [37]. Ischemia has also been shown to induce excitotoxic damage to oligodendrocytes and astrocytes through AMPA receptors [29,38].
- In conclusion, cerebral WM is highly vulnerable to the effects of focal ischemia, among which myelinated axon is damaged first. Early and persistent increase of GFAP immunopositivity in astrocytes might be important for investigating the molecular mechanism of WM infarcts as well as the endogenous or exogenous neuroprotective responses involved. Future studies will be focused on biologic interactive mechanisms among neurons, myelinated axons, astrocytes, oligodendrocytes, and vascular endothelial cells in the experimental model.
DISCUSSION
Acknowledgments
|
Time of post-ischemia |
||||||||
|---|---|---|---|---|---|---|---|---|
| 3 hr | 6 hr | 12 hr | 1 day | 4 days | 7 days | 14 days | 21 days | |
| Light microscopy | ||||||||
| Stroke lesion group | 2 | 2 | 2 | 4 | 4 | 4 | a8a | a8a |
| Sham operation group | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Electron microscopy | 1 | 1 | 1 | 2 | 2 | 2 | 2 | 2 |
| Infarct volume | - | - | - | - | - | - | 14b | - |
- 1. Schneider AT, Kissela B, Woo D, et al. Ischemic stroke subtypes: a population-based study of incidence rates among blacks and whites. Stroke 2004; 35: 1552-6. ArticlePubMed
- 2. Hachinski VC, Potter P, Merskey H. Leuko-araiosis. Arch Neurol 1987; 44: 21-3. ArticlePubMed
- 3. Matsusue E, Sugihara S, Fujii S, Ohama E, Kinoshita T, Ogawa T. White matter changes in elderly people: MR-pathologic correlations. Magn Reson Med Sci 2006; 5: 99-104. ArticlePubMed
- 4. Fu JH, Lu CZ, Hong Z, Dong Q, Luo Y, Wong KS. Extent of white matter lesions is related to acute subcortical infarcts and predicts further stroke risk in patients with first ever ischaemic stroke. J Neurol Neurosurg Psychiatry 2005; 76: 793-6. ArticlePubMedPMC
- 5. Kim GM, Park KY, Avery R, et al. Extensive leukoaraiosis is associated with high early risk of recurrence after ischemic stroke. Stroke 2014; 45: 479-85. ArticlePubMed
- 6. Bailey EL, McCulloch J, Sudlow C, Wardlaw JM. Potential animal models of lacunar stroke: a systematic review. Stroke 2009; 40: e451-8. PubMed
- 7. He SQ, Dum RP, Strick PL. Topographic organization of corticospinal projections from the frontal lobe: motor areas on the lateral surface of the hemisphere. J Neurosci 1993; 13: 952-80. ArticlePubMedPMC
- 8. Lee MC, Jin CY, Kim HS, et al. Stem cell dynamics in an experimental model of stroke. Chonnam Med J 2011; 47: 90-8. ArticlePubMedPMC
- 9. Song S, Park JT, Na JY, et al. Early expressions of hypoxia-inducible factor 1alpha and vascular endothelial growth factor increase the neuronal plasticity of activated endogenous neural stem cells after focal cerebral ischemia. Neural Regen Res 2014; 9: 912-8. ArticlePubMedPMC
- 10. Jang JW, Lee JK, Lee MC, Piao MS, Kim SH, Kim HS. Melatonin reduced the elevated matrix metalloproteinase-9 level in a rat photothrombotic stroke model. J Neurol Sci 2012; 323: 221-7. ArticlePubMed
- 11. Liebigt S, Schlegel N, Oberland J, Witte OW, Redecker C, Keiner S. Effects of rehabilitative training and anti-inflammatory treatment on functional recovery and cellular reorganization following stroke. Exp Neurol 2012; 233: 776-82. ArticlePubMed
- 12. Bush EC, Allman JM. The scaling of white matter to gray matter in cerebellum and neocortex. Brain Behav Evol 2003; 61: 1-5. ArticlePubMedPDF
- 13. Kudo T, Takeda M, Tanimukai S, Nishimura T. Neuropathologic changes in the gerbil brain after chronic hypoperfusion. Stroke 1993; 24: 259-64. ArticlePubMed
- 14. He Z, Yamawaki T, Yang S, Day AL, Simpkins JW, Naritomi H. Experimental model of small deep infarcts involving the hypothalamus in rats: changes in body temperature and postural reflex. Stroke 1999; 30: 2743-51. ArticlePubMed
- 15. Hughes PM, Anthony DC, Ruddin M, et al. Focal lesions in the rat central nervous system induced by endothelin-1. J Neuropathol Exp Neurol 2003; 62: 1276-86. ArticlePubMed
- 16. Frost SB, Barbay S, Mumert ML, Stowe AM, Nudo RJ. An animal model of capsular infarct: endothelin-1 injections in the rat. Behav Brain Res 2006; 169: 206-11. ArticlePubMed
- 17. Whitehead SN, Hachinski VC, Cechetto DF. Interaction between a rat model of cerebral ischemia and beta-amyloid toxicity: inflammatory responses. Stroke 2005; 36: 107-12. ArticlePubMed
- 18. Souza-Rodrigues RD, Costa AM, Lima RR, Dos Santos CD, Picanco-Diniz CW, Gomes-Leal W. Inflammatory response and white matter damage after microinjections of endothelin-1 into the rat striatum. Brain Res 2008; 1200: 78-88. ArticlePubMed
- 19. Tanaka Y, Imai H, Konno K, et al. Experimental model of lacunar infarction in the gyrencephalic brain of the miniature pig: neurological assessment and histological, immunohistochemical, and physiological evaluation of dynamic corticospinal tract deformation. Stroke 2008; 39: 205-12. ArticlePubMed
- 20. Shibata M, Ohtani R, Ihara M, Tomimoto H. White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion. Stroke 2004; 35: 2598-603. ArticlePubMed
- 21. Kim HS, Kim D, Kim RG, et al. A rat model of photothrombotic capsular infarct with a marked motor deficit: a behavioral, histologic, and microPET study. J Cereb Blood Flow Metab 2014; 34: 683-9. ArticlePubMedPMCPDF
- 22. Kim HS, Park MS, Lee JK, Kim HJ, Park JT, Lee MC. Time point expression of apoptosis regulatory proteins in a photochemically-induced focal cerebral ischemic rat brain. Chonnam Med J 2011; 47: 144-9. ArticlePubMedPMC
- 23. Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab 1990; 10: 290-3. ArticlePubMedPDF
- 24. Lee KH, Kang KJ, Moon KS, et al. Prognostic significance of neuronal marker expression in glioblastomas. Childs Nerv Syst 2012; 28: 1879-86. ArticlePubMedPDF
- 25. Kim JM, Lee TH, Lee MC, et al. Endoneurial microangiopathy of sural nerve in experimental vacor-induced diabetes. Ultrastruct Pathol 2002; 26: 393-401. ArticlePubMed
- 26. Donovan V, Kim C, Anugerah AK, et al. Repeated mild traumatic brain injury results in long-term white-matter disruption. J Cereb Blood Flow Metab 2014; 34: 715-23. ArticlePubMedPMCPDF
- 27. Bashkatov AN, Genina EA, Tuchin VV. Tissue optical properties. In: Boas DA, Pitris C, Ramanujam N, eds. Handbook of biomedical optics. Boca Raton: CRC Press, 2011; 67-100.
- 28. Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci 2004; 5: 146-56. ArticlePubMedPDF
- 29. Franke H, Verkhratsky A, Burnstock G, Illes P. Pathophysiology of astroglial purinergic signalling. Purinergic Signal 2012; 8: 629-57. ArticlePubMedPMCPDF
- 30. Sato Y, Chin Y, Kato T, et al. White matter activated glial cells produce BDNF in a stroke model of monkeys. Neurosci Res 2009; 65: 71-8. ArticlePubMed
- 31. Carrera E, Tononi G. Diaschisis: past, present, future. Brain 2014; 137(Pt 9): 2408-22. ArticlePubMed
- 32. Fulmer CG, VonDran MW, Stillman AA, Huang Y, Hempstead BL, Dreyfus CF. Astrocyte-derived BDNF supports myelin protein synthesis after cuprizone-induced demyelination. J Neurosci 2014; 34: 8186-96. ArticlePubMedPMC
- 33. Miyamoto N, Maki T, Shindo A, et al. Astrocytes promote oligodendrogenesis after white matter damage via brain-derived neurotrophic factor. J Neurosci 2015; 35: 14002-8. ArticlePubMedPMC
- 34. Ramos-Cejudo J, Gutiérrez-Fernández M, Otero-Ortega L, et al. Brain-derived neurotrophic factor administration mediated oligodendrocyte differentiation and myelin formation in subcortical ischemic stroke. Stroke 2015; 46: 221-8. ArticlePubMed
- 35. Sozmen EG, Kolekar A, Havton LA, Carmichael ST. A white matter stroke model in the mouse: axonal damage, progenitor responses and MRI correlates. J Neurosci Methods 2009; 180: 261-72. ArticlePubMedPMC
- 36. He Z, Yang SH, Naritomi H, et al. Definition of the anterior choroidal artery territory in rats using intraluminal occluding technique. J Neurol Sci 2000; 182: 16-28. ArticlePubMed
- 37. Karadottir R, Cavelier P, Bergersen LH, Attwell D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 2005; 438: 1162-6. ArticlePubMedPMCPDF
- 38. Bakiri Y, Hamilton NB, Káradóttir R, Attwell D. Testing NMDA receptor block as a therapeutic strategy for reducing ischaemic damage to CNS white matter. Glia 2008; 56: 233-40. ArticlePubMedPMC
REFERENCES
Figure & Data
References
Citations
- Neuroglia and immune cells play different roles in neuroinflammation and neuroimmune response in post-stroke neural injury and repair
Hui Guo, Wen-cao Liu, Yan-yun Sun, Xin-chun Jin, Pan-pan Geng
Acta Pharmacologica Sinica.2026; 47(2): 273. CrossRef - Animal models of focal ischemic stroke: brain size matters
Blazej Nowak, Piotr Rogujski, Raphael Guzman, Piotr Walczak, Anna Andrzejewska, Miroslaw Janowski
Frontiers in Stroke.2023;[Epub] CrossRef - Motor Cortex Plasticity During Functional Recovery Following Brain Damage
Noriyuki Higo
Journal of Robotics and Mechatronics.2022; 34(4): 700. CrossRef - Neurodegeneration, Myelin Loss and Glial Response in the Three-Vessel Global Ischemia Model in Rat
Tatiana Anan’ina, Alena Kisel, Marina Kudabaeva, Galina Chernysheva, Vera Smolyakova, Konstantin Usov, Elena Krutenkova, Mark Plotnikov, Marina Khodanovich
International Journal of Molecular Sciences.2020; 21(17): 6246. CrossRef - Quantitative assessment of demyelination in ischemic stroke in vivo using macromolecular proton fraction mapping
Marina Y Khodanovich, Alena A Kisel, Andrey E Akulov, Dmitriy N Atochin, Marina S Kudabaeva, Valentina Y Glazacheva, Michael V Svetlik, Yana A Medvednikova, Lilia R Mustafina, Vasily L Yarnykh
Journal of Cerebral Blood Flow & Metabolism.2018; 38(5): 919. CrossRef - Immunosignals of Oligodendrocyte Markers and Myelin-Associated Proteins Are Critically Affected after Experimental Stroke in Wild-Type and Alzheimer Modeling Mice of Different Ages
Dominik Michalski, Anna L. Keck, Jens Grosche, Henrik Martens, Wolfgang Härtig
Frontiers in Cellular Neuroscience.2018;[Epub] CrossRef - Administration of Downstream ApoE Attenuates the Adverse Effect of Brain ABCA1 Deficiency on Stroke
Xiaohui Wang, Rongwen Li, Alex Zacharek, Julie Landschoot-Ward, Fengjie Wang, Kuan-Han Hank Wu, Michael Chopp, Jieli Chen, Xu Cui
International Journal of Molecular Sciences.2018; 19(11): 3368. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
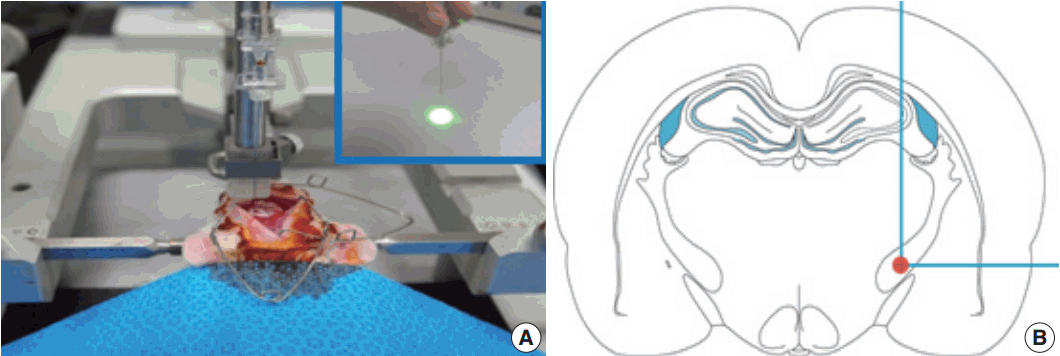
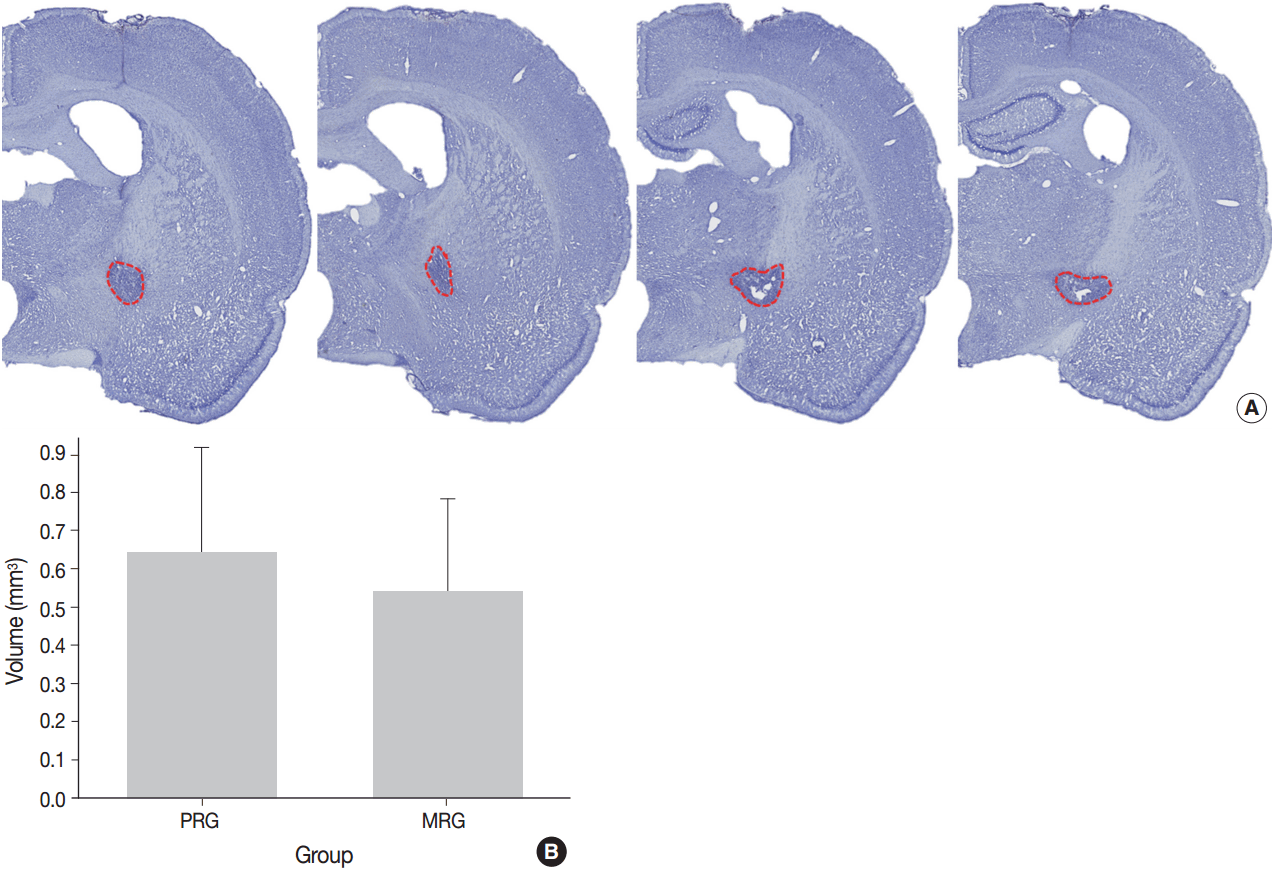
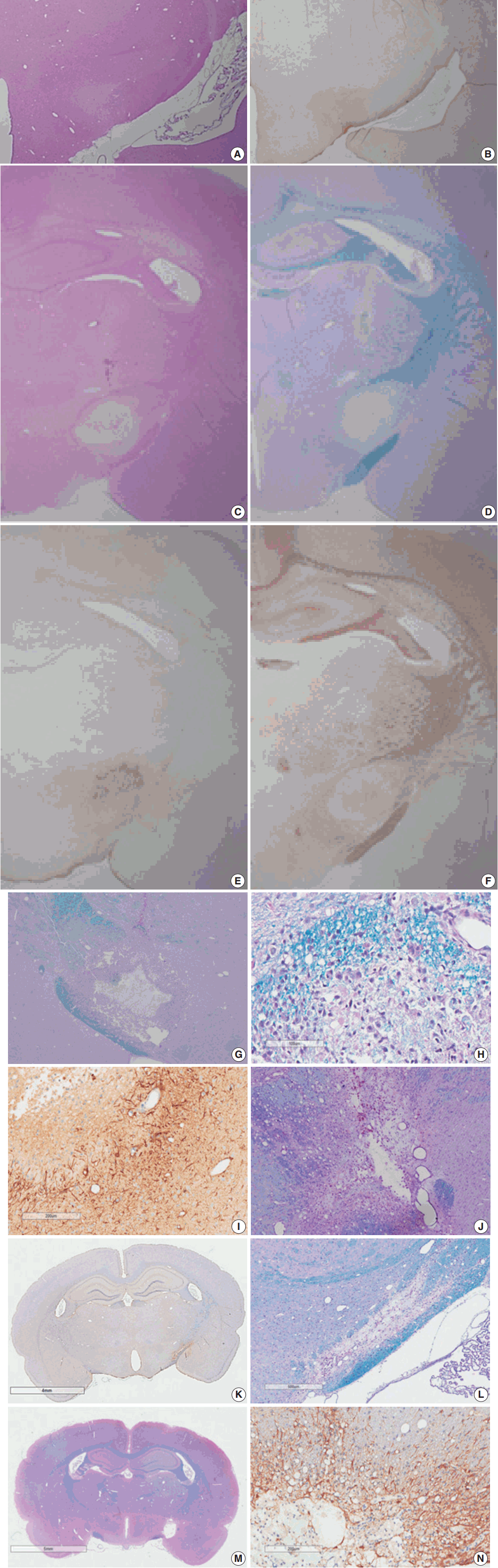
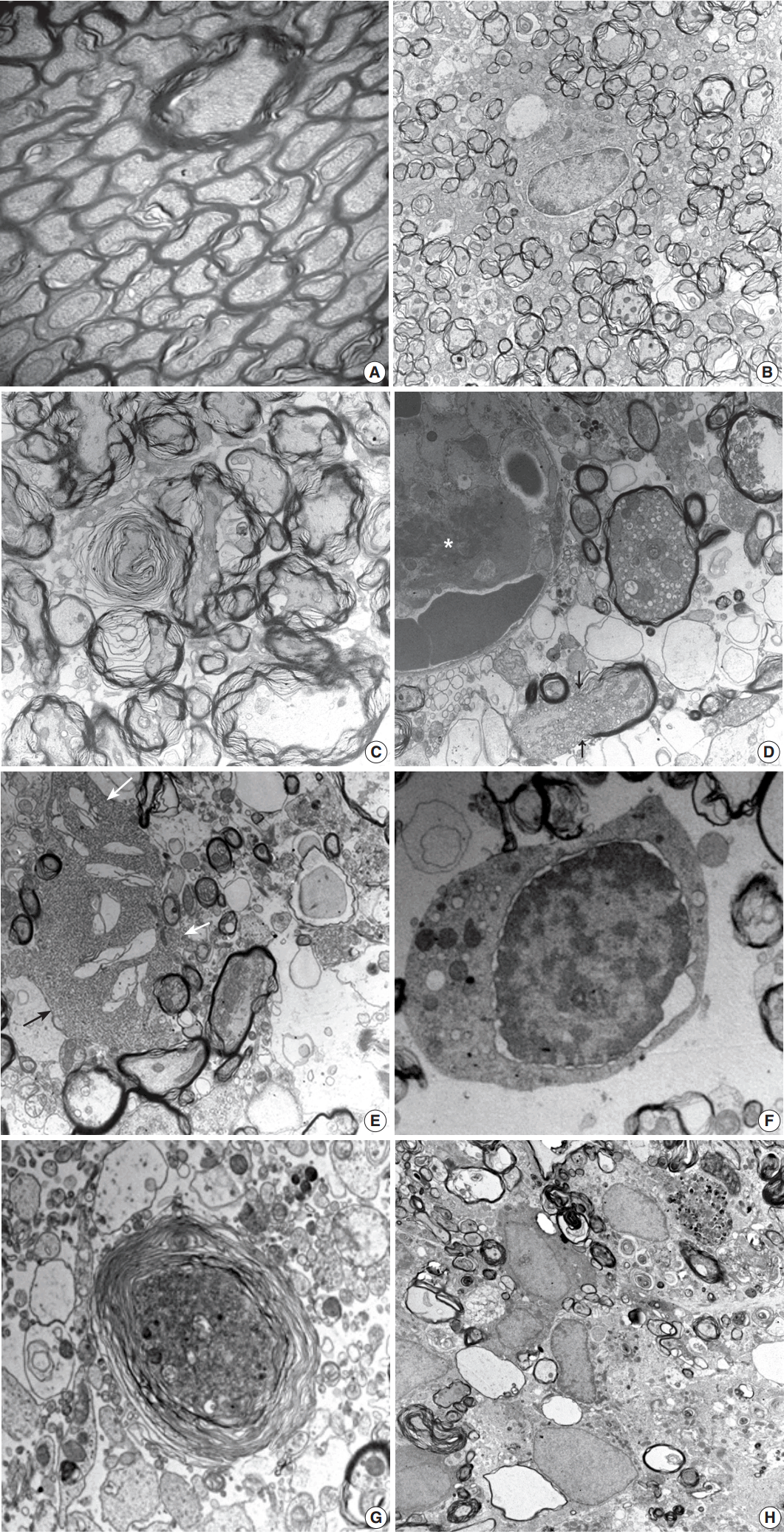




Fig. 1.
Fig. 2.
Fig. 3.
Fig. 4.
| Time of post-ischemia |
||||||||
|---|---|---|---|---|---|---|---|---|
| 3 hr | 6 hr | 12 hr | 1 day | 4 days | 7 days | 14 days | 21 days | |
| Light microscopy | ||||||||
| Stroke lesion group | 2 | 2 | 2 | 4 | 4 | 4 | a8 |
a8 |
| Sham operation group | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Electron microscopy | 1 | 1 | 1 | 2 | 2 | 2 | 2 | 2 |
| Infarct volume | - | - | - | - | - | - | 14 |
- |
| Time (post-ischemia) | Light microscopy | Electron microscopy |
|---|---|---|
| 3-12 hr | Focal lesion with blurred margin | Nodular swelling and loosening of compact myelin sheath |
| Myelin pallor by H&E and LBPAS stain | Submyelin edema, interstitial | |
| Increased GFAP and decreased NFP immunoreactivity from lesion center | Mild wrinkling of axons, but intact neurofilaments and mitochondria | |
| Reactive astrocytes with hypertrophic processes | ||
| 1 day | Focal lesion on PUC with well-defined margin | Fibrin thrombosis and capillary luminal obstruction |
| Increased GFAP immunopositivity in the lesion and surrounding white matter | Axonal swelling and flooding of organelles, extracellular edema | |
| Rupture of myelin sheath with subsequent demyelination | ||
| Loss of NFP immunopositivity and LBPAS stainability | Reactive astrocytes with hypertrophic processes | |
| No inflammatory cell infiltration | Degeneration and apoptosis of oligodendrocytes | |
| 4 days | Focal infarct with central necrotic cavity surrounded by macrophages engulfing myelin debris | Markedly swollen axons with demyelination forming inflated ball or cystic appearance |
| Loss of myelinated axons of PLIC | Marked extracellular edema with exudation | |
| GFAP immunopositive hypertrophic astrocytes along the border of infarct and its vicinity | Necrosis of cellular elements | |
| Macrophages phagocytized with myelin debris and dense bodies | ||
| 7 days | Focal infarct with necrotic cavity surrounded by macrophages engulfing myelin debris, newly formed capillaries, and reactive astrogliosis | Reduced exudative change |
| Degeneration and loss of myelin sheath, demyelinated axons | ||
| Macrophages phagocytized with myelin debris and dense bodies | ||
| 14 days | Focal infarct with necrotic cavity containing macrophages, hypertrophic astrocytes and newly formed capillaris | Demyelinated axons |
| Astrogliosis | ||
| Macrophages, progressively reduced in number | Macrophages, markedly reduced phagocytic debris | |
| 21 days | Focal infarct with reduced necrotic cavity containing few macrophages and angiogliosis | Demyelinated axons |
| Astrogliosis | ||
| Mecrophages with phagocytic debris still present |
Four rats each for the moderate recovery group (MRG) and poor recovery group (PRG); Seven rats each MRG and PRG for measurement of lesion volume.
H&E, hematoxylin and eosin; LBPAS, luxol fast blue–periodic acid-Schiff; GFAP, glial fibrillary acidic protein; NFP, neurofilament protein; PLIC, posterior limb of internal capsule.

