 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 56(4); 2022 > Article
-
Original Article
Founder BRCA1 mutations in Nepalese population -
Anurag Mehta1
 , Himanshi Diwan1, Garima Gupta2, Shrinidhi Nathany1, Shalini Agnihotri3, Surender Dhanda1
, Himanshi Diwan1, Garima Gupta2, Shrinidhi Nathany1, Shalini Agnihotri3, Surender Dhanda1 -
Journal of Pathology and Translational Medicine 2022;56(4):212-216.
DOI: https://doi.org/10.4132/jptm.2022.05.02
Published online: June 15, 2022
1Department of Laboratory, Molecular and Transfusion Services, Rajiv Gandhi Cancer Institute and Research Centre (RGCIRC), New Delhi, India
2Research Department, Indian Institute of Technology, New Delhi, India
3Department of Research, Rajiv Gandhi Cancer Institute and Research Centre (RGCIRC), New Delhi, India
- Corresponding Author: Himanshi Diwan, MD, Department of Laboratory, Molecular and Transfusion Services, Rajiv Gandhi Cancer Institute and Research Centre, New Delhi 110085, India, Tel: +91-9413542252, Fax: +91-11-27051037, E-mail: himanshidiwan89@gmail.com
© 2022 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
-
Background
- Founder mutation is a heritable genetic alteration observed with high frequency in a geographically and culturally isolated population where one or more ancestors becomes the forebearer of the altered gene. The current study reports two founder mutations in the BRCA1 gene in the Nepalese people.
-
Methods
- Germline BRCA testing in all surface epithelial ovarian cancers and the selected case of breast, prostate, and pancreatic cancers has been the standard practice from 2016 to 2021. One thousand one hundred thirty-three probands were screened for germline BRCA variants by next generation sequencing. The variants were classified as per the American Society of Medical Genetics and Genomics recommendations. Pathogenic (class V) and likely pathogenic (class IV) were considered clinically relevant and utilized for cascade screening.
-
Results
- Nepalese population made up a subcohort of 5.12% (58/1,133) of probands tested for germline BRCA1/2 variants. Twenty-seven of these 58 tested harbored pathogenic genetic alterations in BRCA1/2 genes, with 23 being BRCA1 mutant. Sixteen of 23 BRCA1 mutant cases shared one common pathogenic mutation c.2214_2215insT (p.Lys739Ter) (NM_007294.4). Additionally, a second highly recurrent mutation in BRCA1 gene c.5068A>T (p.Lys1690Ter) (NM_007294.4) was noted in six patients from this population.
-
Conclusions
- The overwhelming abundance of the above two variants in a geographically confined population confers these two genetic alterations a status of founder mutations amongst the people of Nepal. A more extensive population-based study to reaffirm these findings will help establish a dual site-specific germline testing similar to the “Multisite-3-assay” in Ashkenazi Jews as the primary screening tool, especially in a resource-constrained environment.
- Research setting and subjects
- One thousand one hundred thirty-three patients diagnosed with breast/ovarian/prostate/pancreatic cancer and who fulfilled the BRCA testing criteria recommended by National Comprehensive Cancer Network (NCCN) were included in the study [15]. Each eligible subject was explicitly explained and counseled about the pros and cons of undergoing BRCA testing by the institutional genetic counselor, and informed written consent was obtained for testing and using the information for research. The included participants were screened for the mutations in the BRCA genes by next generation sequencing (NGS), and the large genomic rearrangements (Big Indels) were examined through Multiplex Ligation-dependent Probe Amplification (MLPA).
- Isolation of DNA from blood, NGS, and data analysis
- Genomic DNA was isolated from 2 mL of peripheral blood of the index case using the commercially available DNA isolation kit (Qiagen DNeasy Blood and Tissue kit, Qiagen NV, Hilden, Germany), following the manufacturer’s instructions. Isolated DNA was quantified by Qubit 3.0 Fluorometric quantitation (Thermo Fisher Scientific, Waltham, MA, USA). NGS library was prepared manually with 10 ng of the isolated DNA using Oncomine BRCA assay – A328400 (Thermo Fisher Scientific) as detailed elsewhere [16]. Data generated from the runs was assessed for quality metrics on Torrent Suite Viewer (Ion Torrent Suite 5.10, Thermo Fisher Scientific) for parameters like the number of mapped reads, average base coverage depth, uniformity of coverage, coverage at 1 ×, 20 ×, and 100 ×, strand bias, end to end amplicon reads. Thresholds were employed as enunciated in The National Cancer Institute-Molecular Analysis for Therapy Choice (NCI-MATCH) trial [17]. Mapped reads of > 1,00,000 with 90% uniformity, average base coverage depth ≥ 100, and > 90% coverage at 100× were considered optimal for reporting though the base coverage and mapped reads were usually far more. The variants were classified according to the American Society of Medical Genetics and Genomics recommendations for standards of interpretation and reporting of sequence variations [18].
- Cases tested negative for BRCA1/BRCA2 mutations were further investigated for possible large genomic rearrangements (Big Indels) by MLPA assay described previously elsewhere [16].
- Statistical analysis
- Descriptive statistics were used to summarize the data. The data were analyzed statistically using SPSS ver. 23.0 (IBM Corp., Armonk, NY, USA). The statistical analysis comprised of calculating means and proportions. Appropriate tests of significance were applied, and a p-value of < .05 was considered significant.
MATERIALS AND METHODS
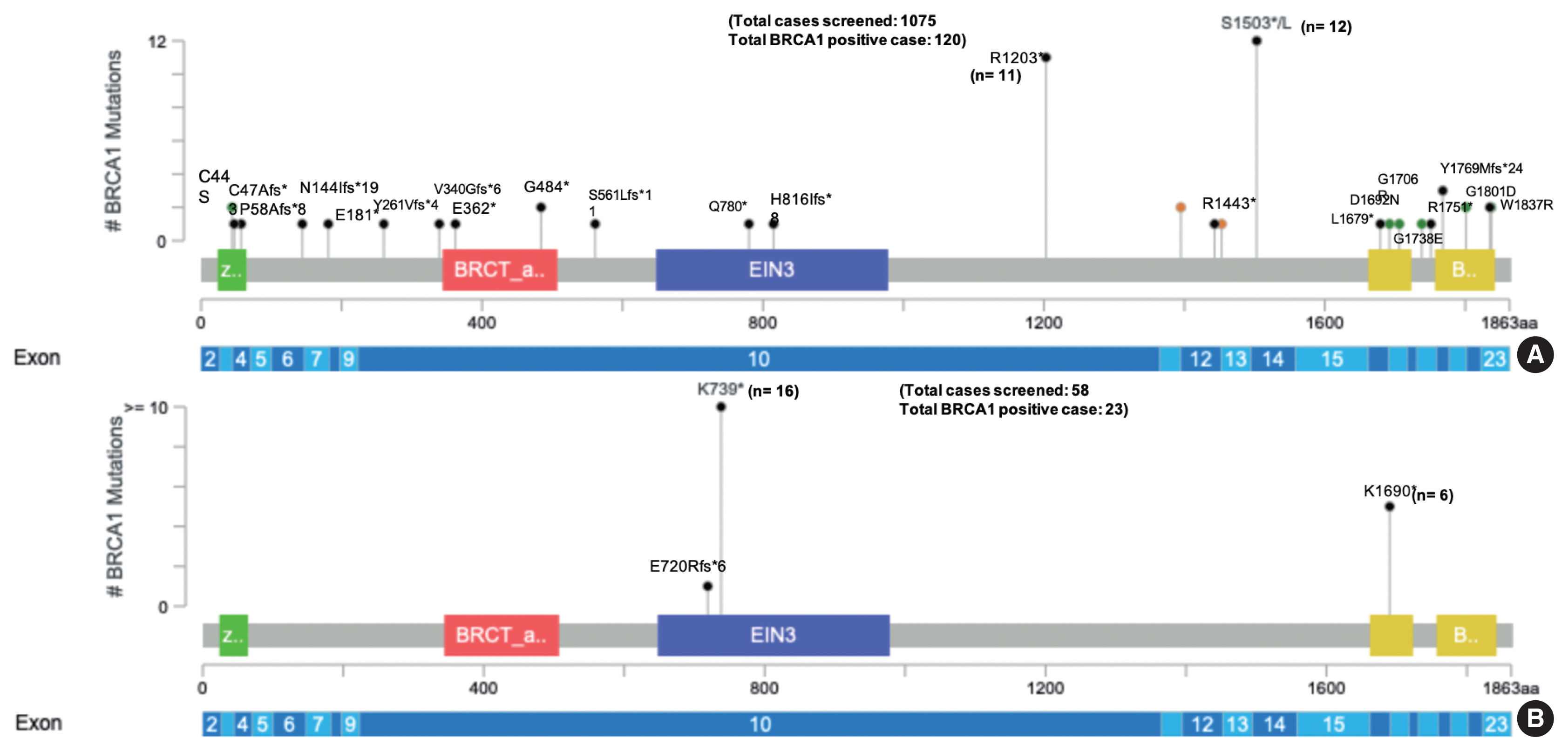
- One thousand one hundred thirty-three comprehensive BRCA1 and BRCA2 testing for different ethnic populations, comprising the Indian, Nepalese, Bangladeshi, Afghani, Kenyan, Iraqi, and the Myanmarese, were performed at the Molecular Diagnostic laboratory of a tertiary cancer care hospital in India. Of the 1,133 patients tested, 58 were Nepalese descent (5.1%) with a mean age of 47.3 years. Twenty-seven of the 58 Nepalese patients harbored pathogenic BRCA1/BRCA2 genetic alteration, 16 breast, and 11 ovarian carcinoma cases. The mean age was 46.5 years in the Nepalese against 46.8 years in the rest of the cohort (95% confidence interval, −4.288 to 5.088; p = .867). Strikingly 23 of these 27 affected probands carried BRCA1 mutation and, far more significantly, just two recurrent nonsense mutations, as shown in Table 1 below. Only one proband out of 23 with BRCA1 inactivating alteration had a frameshift mutation outside these two genetic alterations. The two recurrent BRCA1 alterations (c.2214_2215insT and c.5068A>T) were not found in any other ethnic population in the study (Fig. 1).
- A BRCA mutation is seen in 27 Nepalese patients (27/58), accounting for 46.5% of tested patients. In comparison, BRCA mutation is observed in 227 non-Nepalese patients (227/1,075), accounting for 21.1%. The preceding emphasizes that BRCA mutation is significantly higher in the Nepalese population than in the non-Nepalese population (Fisher exact test, statistical value is <0.001). Also, BRCA1 mutation was more common in Nepalese subcohort (23/58) than non-Nepalese subcohort (167/1,075) (Fisher exact test, statistical value is <0.001).
- Of the total BRCA mutant cases, c.2214_2215insT is the most frequently observed BRCA1 variant in the Nepalese population (16/27), constituting 59.2% of the total BRCA mutant cases followed by c.5068A>T (6/27) (22.2%). Thirty three point three three percentage of these 27 BRCA mutant cases constituted triple-negative breast carcinoma with 29.6% luminal type breast cancer and 25.9% high-grade serous ovarian carcinoma, as summarized in Table 2. Only 11 (11/27) BRCA mutant cases had a family history of breast/ovarian cancer.
- One of the probands was tested for somatic alteration in the BRCA1 gene in ovarian carcinoma, and p.Lys1690Ter was detected in the BRCA1 gene at a high variant allele frequency (VAF). Combined with our observation of this variant being highly repetitive in the Nepalese people and the high VAF, a peripheral blood testing was done by an orthogonal method (site-specific Sanger sequencing) and confirmed as germline.
RESULTS
- BRCA1 and BRCA2 mutations show variable prevalence in different ethnic populations. The prevalence of BRCA1/2 mutation in the general population is estimated to be around 1/800 to 1/1,000 or at the rate of 0.125 to 0.1%. In contrast, the prevalence increases several-fold in the geographically confined ethnic groups with founder mutation as in Ashkenazi Jews, where BRCA mutation is observed at a frequency of 2.5% [12,13]. The present study highlighted a similarly high incidence of BRCA mutation in the Nepalese patient cohort (46.5%), with the BRCA1 gene altered in 39.6% and the BRCA2 in 7%.
- The discovery of a founder mutation in Ashkenazi Jews population paved the way to the “Multisite 3 assay” testing for BRCA1 185delAG, BRCA1 5382insC, and BRCA2 6174delT. The present study identifies two founder mutations in the BRCA1 gene (c.2214_2215insT and c.5068A>T) in the Nepalese population. Significantly, none from the non-Nepalese cohort of the 1,075 probands tested positive for these two mutations. This exclusiveness and high frequency of just two variants in an ethnic group support the founder nature of these variants.
- These two alterations account for 85.2% of all cases detected with germline BRCA mutation in the Nepalese subcohort, which is far higher than the three mutations that make up 73.2% of all germline BRCA mutations amongst Ashkenazi Jews [21]. Such high occurrence of just two mutations opens up the possibility that, like in Ashkenazi Jews, the first tier of testing can be reduced to dual site-specific testing for BRCA1 c.2214_2215insT and BRCA1 c.5068A>T for the Nepalese people. Failing to identify one of the two nonsense mutations shall prompt further testing by a more extensive NGS-based assay.
- One of the index cases of ovarian carcinoma screened for somatic BRCA testing had c. 5068A>T mutation in BRCA1 gene at a high VAF and confirmed as germline mutation on orthogonal testing by site-specific Sanger sequencing on peripheral blood. Further, even when detected in tumor tissue, these two mutations shall get full consideration of germline origin, and germline testing should follow.
- A positive family history of breast/ovarian carcinoma was found in 40.71% of BRCA mutant cases in the Nepalese subcohort, clearly far more common than non-Nepalese subcohort (23.8%). The observation of a significantly high BRCA mutant population sans a family history of cancer predisposition thus mandates testing all patients for BRCA irrespective of the family history as emphasized by the NCCN guidelines [15].
- c.2214_2215insT is located at chr17:41245334; exon 10 of 24 results in a frameshift that generates a new stop codon in place of Lysine, either causing the formation of a truncated BRCA1 protein or lack of it due to Nonsense-mediated mRNA decay [22,23]. Likewise, c.5068A>T is located at chr17:41219631; exon 17 of 23; position 57 of 78 (on assembly GRCh37) [22] also forms a premature stop codon in place of Lysine with similar effects as for the other founder mutation. These mutations were reported by the first author in the LOVD Database [24]. The variants above are not reported in the population databases (ExAC) [25]. The Clinvar has annotated these variants to be pathogenic [26].
- The present hospital-based study on cancer patients acting as probands reveals founder mutations in the Nepalese people. A more extensive population-based study confirming their high-frequency and lack of other randomly distributed deleterious mutations in BRCA genes shall permit the development of a dual site-specific BRCA1 testing as the first-line screen for probands and kindred. Such a strategy shall prevent the need for NGS, drastically reduce the cost of BRCA testing, allow deeper reach, especially in a resource-constrained setting. This strategy shall also provide a fillip to a preventive approach towards cancer.
- BRCA1 c.2214_2215insT and BRCA1 c. 5068A>T are the two germline alterations in the Nepalese population with founder effect and incidence of 85.2%. Further confirmation of our findings by a more extensive population-based study shall allow developing a limited dual site-specific germline test as a first-line screening tool, especially in a resource-restricted environment with limited accessibility to NGS. Additionally, Founder mutations can help conclude or rule out an anthropological hypothesis and has immense research potential in that field.
DISCUSSION
Ethics Statement
This study was approved by the institutional review board (Rajiv Gandhi Cancer Institute and Research Center), vide the ethical approval letter number RGCIRC/IRB-BHR/41/2020. The study was conducted per the Declaration of Helsinki. Informed written consent was obtained for testing and using the information for research.
Availability of Data and Material
The datasets generated or analyzed during the study are available from the corresponding author on reasonable request.
Code Availability
Not applicable.
Author Contributions
Conceptualization: AM. Data curation: GG, HD, SA, SN. Formal analysis: AM, GG, HD, SN. Investigation: AM, HD, GG, SN, SA, SD. Methodology: AM. Project administration: AM. Resources: SD. Supervision: AM. Visualization: HD, SN. Writing—original draft: GG, HD. Writing—review & editing: AM.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
Funding Statement
No funding to declare.
| BRCA mutant Nepalese cohort (n = 27) | BRCA wild Nepalese cohort (n = 31) | BRCA mutant non-Nepalese subcohort (n = 227) | BRCA wild non-Nepalese cohort (n = 848) | |
|---|---|---|---|---|
| Age (yr) | ||||
| Meana | 46.5 | 51.6 | 46.8 | 51.6 |
| Median (range) | 44.5 (32–76) | 52 (27–72) | 44.5 (29–66) | 52 (29–86) |
| Ovarian carcinoma with the morphology of HGSC | 7 (25.9) | 10 (32.5) | 90 (39.6) | 160 (18.8) |
| TNBC cases | 9 (33.3) | 11 (35.5) | 96 (42.3) | 300 (35.4) |
| Luminal type breast carcinoma cases | 8 (29.6) | 6 (19.3) | 23 (10.1) | 207 (24.4) |
| HER-2 enriched breast carcinoma | 1 (3.7) | 1 (3.2) | 2 (0.1) | 26 (3.1) |
| Cases with ovarian HGSC and TNBC | 1 (3.7) | 0 | 10 (4.4) | 2 (0.02) |
| Cases with endometrioid ovarian carcinoma and TNBC | 0 | 0 | 0 | 2 (0.02) |
| Cases with mixed ovarian carcinoma | 0 | 0 | 2 (0.1) | 0 |
| Cases with ovarian low-grade serous carcinoma | 0 | 0 | 0 | 2 (0.02) |
| Cases with ovarian endometrioid carcinoma | 0 | 2 (6.4) | 0 | 5 (0.6) |
| Prostate carcinoma cases | 0 | 1 (3.2) | 2 (0.1) | 62 (7.3) |
| Pancreatic carcinoma cases | 0 | 0 | 2 (0.1) | 82 (9.7) |
| Cases with positive family history | 11 (40.71) | 4 (12.9) | 54 (23.8) | 43 (5.1) |
- 1. Kuchenbaecker KB, Hopper JL, Barnes DR, et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 2017; 317: 2402-16. PubMed
- 2. Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003; 72: 1117-30. ArticlePubMedPMC
- 3. Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol 2007; 25: 1329-33. ArticlePubMedPMC
- 4. Mehta A. BRCA1 and BRCA2 mutations in ovarian cancer. J Curr Oncol 2018; 1: 1-4. Article
- 5. Tai YC, Domchek S, Parmigiani G, Chen S. Breast cancer risk among male BRCA1 and BRCA2 mutation carriers. J Natl Cancer Inst 2007; 99: 1811-4. ArticlePubMedPMC
- 6. Levy-Lahad E, Friedman E. Cancer risks among BRCA1 and BRCA2 mutation carriers. Br J Cancer 2007; 96: 11-5. ArticlePubMedPMCPDF
- 7. Oh M, Alkhushaym N, Fallatah S, et al. The association of BRCA1 and BRCA2 mutations with prostate cancer risk, frequency, and mortality: a meta-analysis. Prostate 2019; 79: 880-95. ArticlePubMedPDF
- 8. Nyberg T, Frost D, Barrowdale D, et al. Prostate cancer risks for male BRCA1 and BRCA2 mutation carriers: a prospective cohort study. Eur Urol 2020; 77: 24-35. PubMedPMC
- 9. Hu C, Hart SN, Polley EC, et al. Association between inherited germline mutations in cancer predisposition genes and risk of pancreatic cancer. JAMA 2018; 319: 2401-9. ArticlePubMedPMC
- 10. Nelson HD, Fu R, Goddard K, et al. Risk assessment, genetic counseling, and genetic testing for BRCA-related cancer: systematic review to update the U.S. Preventive Services Task Force Recommendation. Report AHRQ Publication No. 12-05164-EF-1. Portland: Oregon Health and Science University, 2013.
- 11. Hall MJ, Reid JE, Burbidge LA, et al. BRCA1 and BRCA2 mutations in women of different ethnicities undergoing testing for hereditary breast-ovarian cancer. Cancer 2009; 115: 2222-33. ArticlePubMedPMC
- 12. Balmana J, Diez O, Rubio IT, Cardoso F; ESMO Guidelines Working Group. BRCA in breast cancer: ESMO clinical practice guidelines. Ann Oncol 2011; 22(Suppl 6): vi31-4. ArticlePubMed
- 13. Rosenthal E, Moyes K, Arnell C, Evans B, Wenstrup RJ. Incidence of BRCA1 and BRCA2 non-founder mutations in patients of Ashkenazi Jewish ancestry. Breast Cancer Res Treat 2015; 149: 223-7. ArticlePubMedPDF
- 14. Yadava YP. Linguistic diversity in Nepal: perspectives on language policy. International Seminar on Constitutionalism and Diversity in Nepal; 2007 Aug 22–24; Kathmandu, Nepal.
- 15. Daly MB, Pilarski R, Yurgelun MB, et al. NCCN guidelines insights: genetic/familial high-risk assessment: breast, ovarian, and pancreatic, version 1.2020. J Natl Compr Canc Netw 2020; 18: 380-91. PubMed
- 16. Mehta A, Vasudevan S, Sharma SK, et al. Germline BRCA1 and BRCA2 deleterious mutations and variants of unknown clinical significance associated with breast/ovarian cancer: a report from North India. Cancer Manag Res 2018; 10: 6505-16. PubMedPMC
- 17. Lih CJ, Harrington RD, Sims DJ, et al. Analytical validation of the next-generation sequencing assay for a nationwide signal-finding clinical trial: molecular analysis for therapy choice clinical trial. J Mol Diagn 2017; 19: 313-27. PubMedPMC
- 18. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405-24. ArticlePubMedPMCPDF
- 19. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6: pl1.ArticlePubMedPMC
- 20. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2: 401-4. ArticlePubMedPMCPDF
- 21. Tonin P, Weber B, Offit K, et al. Frequency of recurrent BRCA1 and BRCA2 mutations in Ashkenazi Jewish breast cancer families. Nat Med 1996; 2: 1179-83. ArticlePubMedPDF
- 22. Kopanos C, Tsiolkas V, Kouris A, et al. VarSome: the human genomic variant search engine. Bioinformatics 2019; 35: 1978-80. ArticlePubMedPDF
- 23. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014; 11: 361-2. ArticlePubMedPDF
- 24. Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat 2011; 32: 557-63. ArticlePubMed
- 25. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020; 581: 434-43. ArticlePubMedPMC
- 26. Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 2018; 46: D1062-7. ArticlePubMedPMC
REFERENCES
Figure & Data
References
Citations
- Hiding in plain sight: a partial deletion of BRCA1 exon 7 undetectable by MLPA is a Nepali founder variant
Virginia Clowes, Jenny C Taylor, Alistair T Pagnamenta
Journal of Medical Genetics.2025; 62(2): 54. CrossRef - Spectrum of BRCA1/2 pathogenic variants in Southern and Western Asia-a systematic review
Samra Khan, Ikram A. Burney, Mahrukh Nasir, Humaira Saleem, Muhammad Irfan, Muhammad Shakeel, Ishtiaq Ahmad Khan
Mutation Research - Reviews in Mutation Research.2025; 796: 108549. CrossRef - Finding significance: New perspectives in variant classification of the RAD51 regulators, BRCA2 and beyond
Hayley L. Rein, Kara A. Bernstein
DNA Repair.2023; 130: 103563. CrossRef - Digital PCR as a Highly Sensitive Diagnostic Tool: A Review
K. V. Kopylova, Ed. W. Kasparov, I. V. Marchenko, M. V. Smolnikova
Molecular Biology.2023; 57(5): 793. CrossRef - Digital PCR as a Highly Sensitive Diagnostic Tool: a Review
K. V. Kopylova, Ed. W. Kasparov, I. V. Marchenko, M. V. Smolnikova
Молекулярная биология.2023; 57(5): 771. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
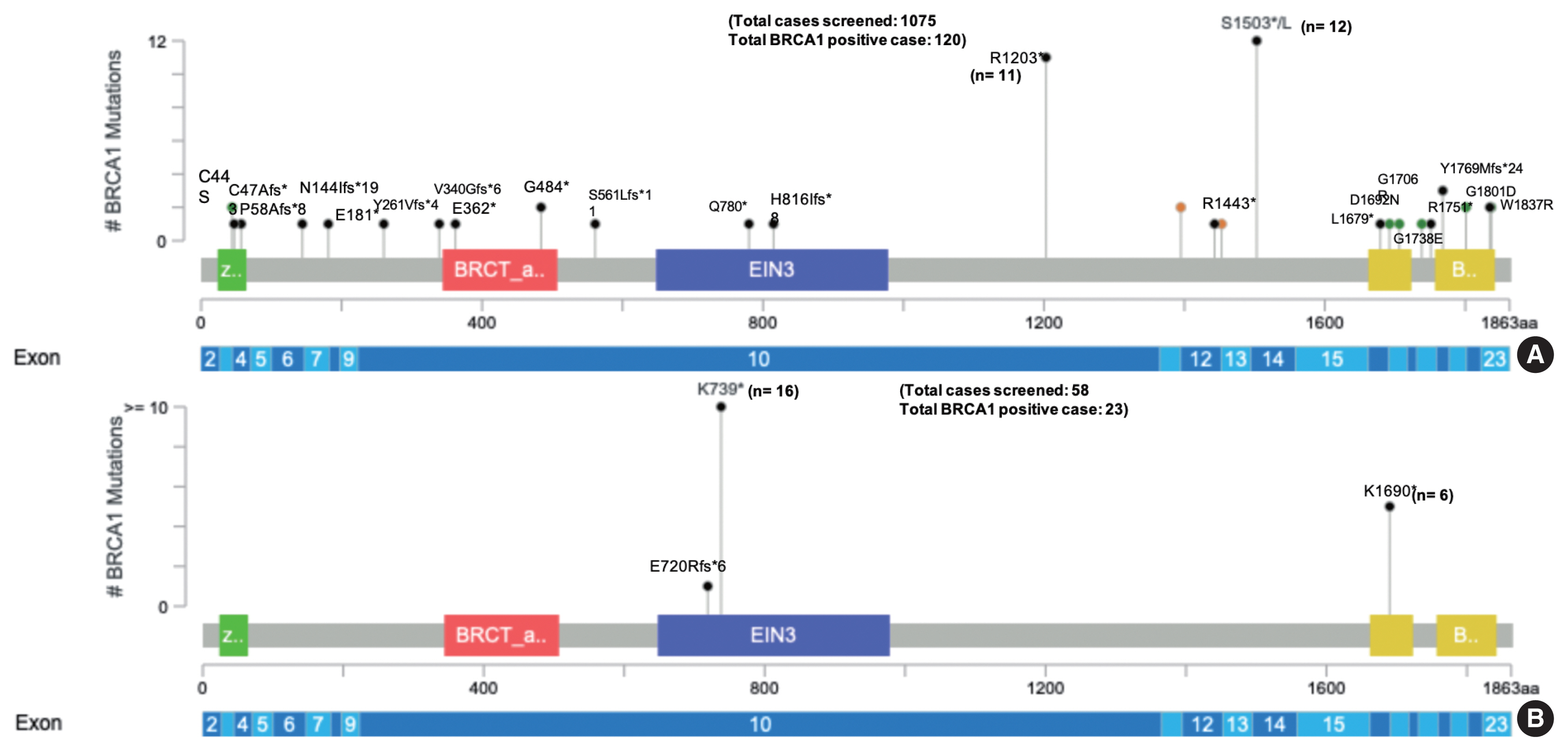

Fig. 1
| Gene | Variant | NM transcript | No. of patients with the variant |
|---|---|---|---|
| BRCA1 | c.2214_2215insT (p.Lys739Ter) | NM_007294.4 | 16 |
| BRCA1 | c.5131A>T(p.Lys1690Ter) | NM_007294.4 | 6 |
| BRCA1 | c.2157_2158insA (p.Glu720ArgfsTer6) | NM_007294.4 | 1 |
| BRCA2 | c.1303_1304insA(p.Arg435fs) | NM_000059.4 | 1 |
| BRCA2 | c.2808_2811 del ACAA (p.Ala938fs) | NM_000059.4 | 1 |
| BRCA2 | c.6270_6271delTA(p.His2090glnfsTer9) | NM_000059.4 | 1 |
| BRCA2 | c.7030delA(p.Ile2344TyrfsTer23 | NM_000059.4 | 1 |
| BRCA mutant Nepalese cohort (n = 27) | BRCA wild Nepalese cohort (n = 31) | BRCA mutant non-Nepalese subcohort (n = 227) | BRCA wild non-Nepalese cohort (n = 848) | |
|---|---|---|---|---|
| Age (yr) | ||||
| Mean |
46.5 | 51.6 | 46.8 | 51.6 |
| Median (range) | 44.5 (32–76) | 52 (27–72) | 44.5 (29–66) | 52 (29–86) |
| Ovarian carcinoma with the morphology of HGSC | 7 (25.9) | 10 (32.5) | 90 (39.6) | 160 (18.8) |
| TNBC cases | 9 (33.3) | 11 (35.5) | 96 (42.3) | 300 (35.4) |
| Luminal type breast carcinoma cases | 8 (29.6) | 6 (19.3) | 23 (10.1) | 207 (24.4) |
| HER-2 enriched breast carcinoma | 1 (3.7) | 1 (3.2) | 2 (0.1) | 26 (3.1) |
| Cases with ovarian HGSC and TNBC | 1 (3.7) | 0 | 10 (4.4) | 2 (0.02) |
| Cases with endometrioid ovarian carcinoma and TNBC | 0 | 0 | 0 | 2 (0.02) |
| Cases with mixed ovarian carcinoma | 0 | 0 | 2 (0.1) | 0 |
| Cases with ovarian low-grade serous carcinoma | 0 | 0 | 0 | 2 (0.02) |
| Cases with ovarian endometrioid carcinoma | 0 | 2 (6.4) | 0 | 5 (0.6) |
| Prostate carcinoma cases | 0 | 1 (3.2) | 2 (0.1) | 62 (7.3) |
| Pancreatic carcinoma cases | 0 | 0 | 2 (0.1) | 82 (9.7) |
| Cases with positive family history | 11 (40.71) | 4 (12.9) | 54 (23.8) | 43 (5.1) |
Values are presented as number (%) unless otherwise indicated. HGSC, high-grade serous carcinoma; TNBC, triple-negative breast carcinoma; HER-2, human epidermal growth factor receptor 2. p = .867; 95% confidence interval, −4.288 to 5.088 (Nepalese vs. non-Nepalese

