 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 56(4); 2022 > Article
-
Case Study
Primary pulmonary epithelioid inflammatory myofibroblastic sarcoma: a rare entity and a literature review -
Priyanka Singh1
 , Aruna Nambirajan1, Manish Kumar Gaur2, Rahul Raj1, Sunil Kumar2, Prabhat Singh Malik3, Deepali Jain1
, Aruna Nambirajan1, Manish Kumar Gaur2, Rahul Raj1, Sunil Kumar2, Prabhat Singh Malik3, Deepali Jain1 -
Journal of Pathology and Translational Medicine 2022;56(4):231-237.
DOI: https://doi.org/10.4132/jptm.2022.05.08
Published online: July 7, 2022
1Department of Pathology, All India Institute of Medical Sciences, New Delhi, India
2Department of Surgical Oncology, Dr. BRA Institute Rotary Cancer Hospital, All India Institute of Medical Sciences, New Delhi, India
3Department of Medical Oncology, Dr. BRA Institute Rotary Cancer Hospital, All India Institute of Medical Sciences, New Delhi, India
- Corresponding Author: Deepali Jain, MD, FIAC, Department of Pathology, All India Institute of Medical Sciences, New Delhi 110029, India Tel: +91-1126549200, E-mail: deepalijain76@gmail.com
© 20222020 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
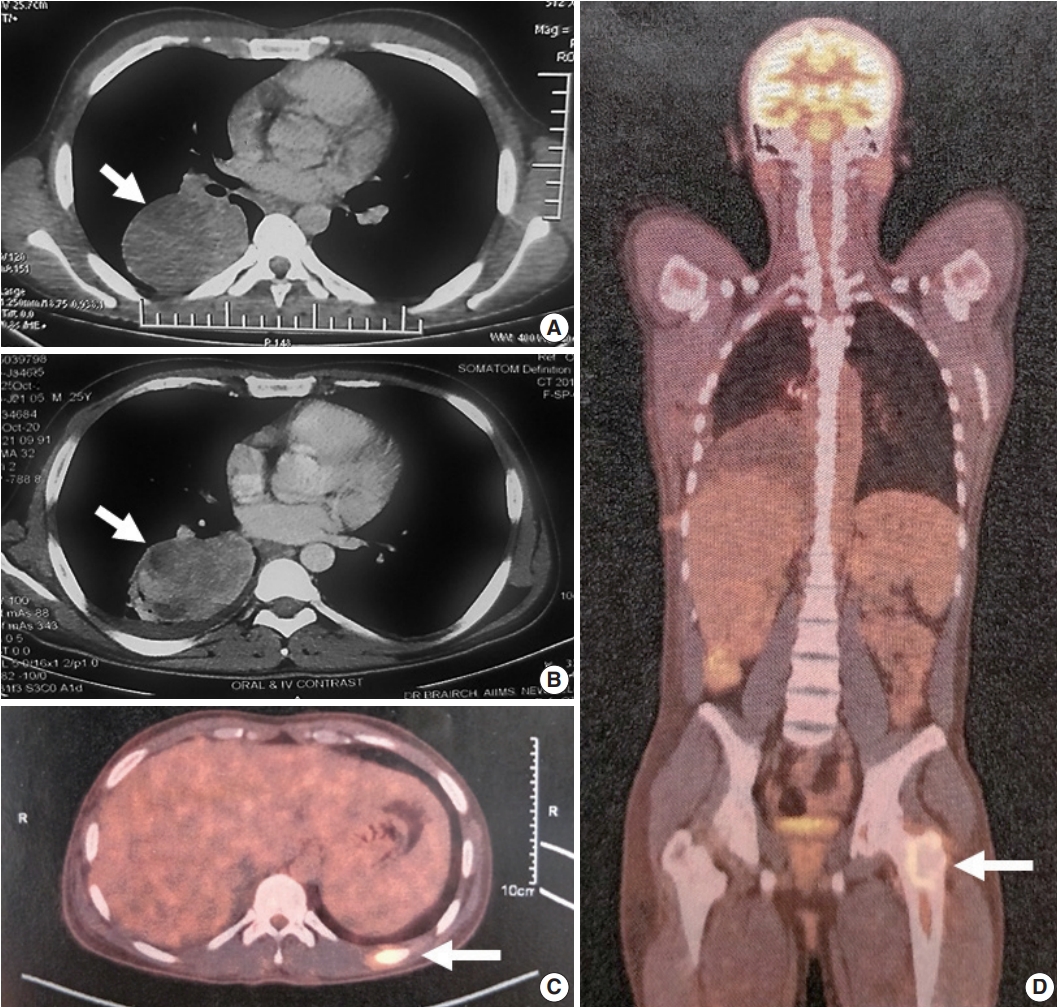
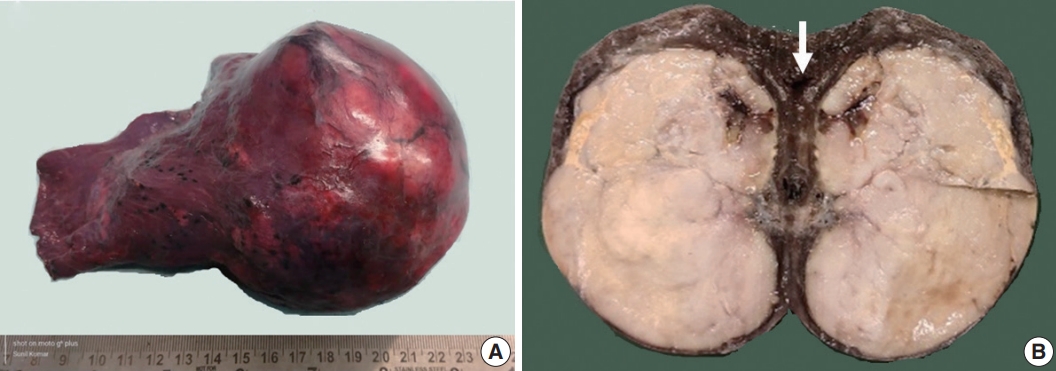
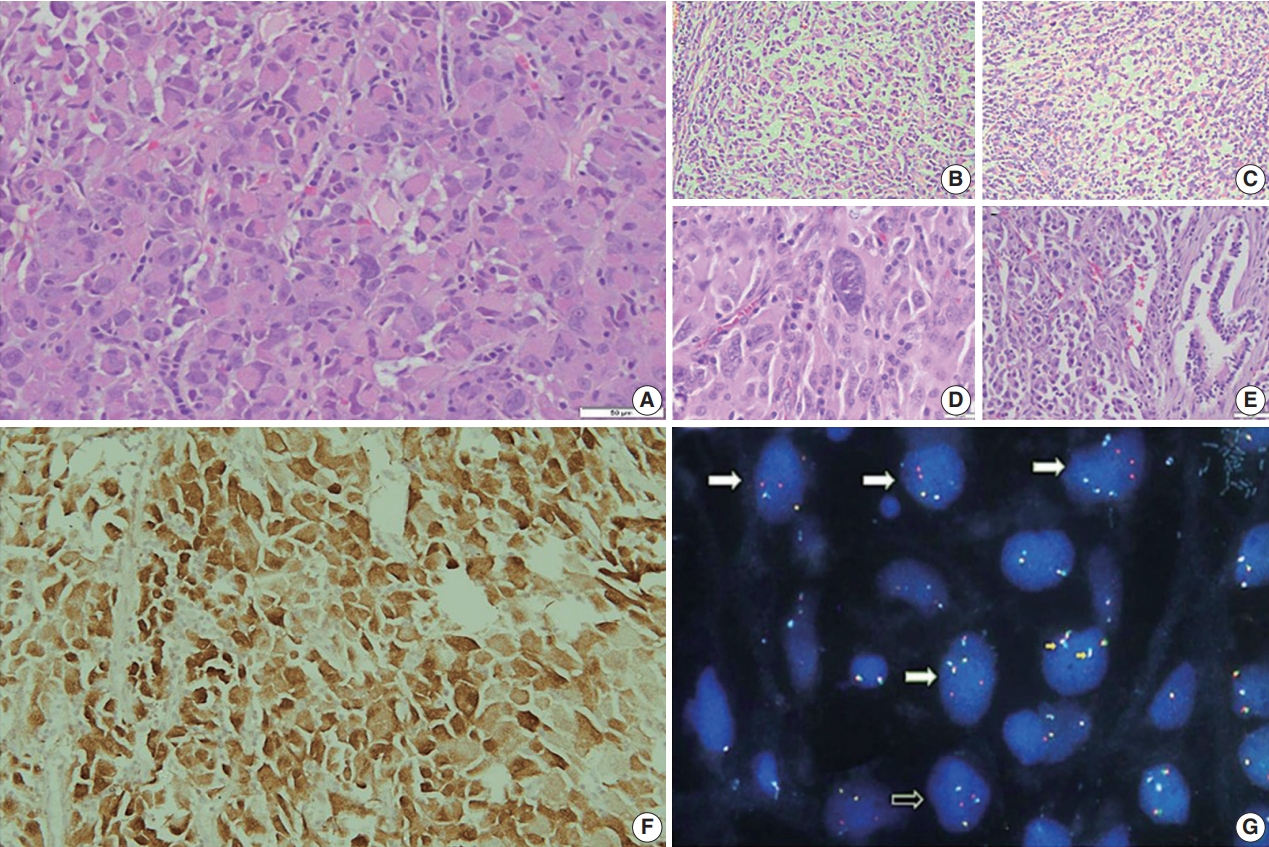
Figure & Data
References
Citations
- Inflammatory Myofibroblastic Tumor: An Updated Review
Joon Hyuk Choi
Cancers.2025; 17(8): 1327. CrossRef - Epithelioid Inflammatory Myofibroblastic Sarcoma: Case Series With a First Report of CLTC::ALK Fusion in an Aggressive Disease
Daisy Maharjan, Carina Dehner, Ali Alani, Robert Bell, Sheila Segura
Genes, Chromosomes and Cancer.2025;[Epub] CrossRef - ALK rearranged malignant mesenchymal neoplasms of thorax: therapeutically targetable ‘ALKomas’ beyond the spectrum of non-small cell lung carcinomas and thoracic inflammatory myofibroblastic tumors
Shreya Sadhu, Adarsh Barwad, Asit Ranjan Mridha, Prabhat Singh Malik, Aruna Nambirajan, Deepali Jain
Virchows Archiv.2025; 487(5): 1003. CrossRef - Mediastinal epithelioid inflammatory myofibroblastic sarcoma with the EML4‐ALK fusion: A case report and literature review
Tingyu Pan, Xinyu Sun, Xiao Wu, Futing Tang, Xianmei Zhou, Qian Wang, Shi Chen
Respirology Case Reports.2024;[Epub] CrossRef - Primary epithelioid inflammatory myofibroblastic sarcoma of the brain with EML4::ALK fusion mimicking intra-axial glioma: a case report and brief literature review
Eric Eunshik Kim, Chul-Kee Park, Koung Mi Kang, Yoonjin Kwak, Sung-Hye Park, Jae-Kyung Won
Journal of Pathology and Translational Medicine.2024; 58(3): 141. CrossRef - Epithelioid Inflammatory Myofibroblastic Sarcoma: A Report of a Rare Case
Varun Ronanki, Vaddatti Tejeswini, Inuganti Venkata Renuka, Shaik Raheema, Bakkamanthala S K Kanth
Cureus.2024;[Epub] CrossRef - Thoracic epithelioid inflammatory myofibroblastic sarcoma: a rare and aggressive disease with case report and literature review
Linke Yang, Pei Li, Runze Liu, Baomin Feng, Huiqing Mao, Xiaoyong Tang, Guangjian Yang
Discover Oncology.2024;[Epub] CrossRef - Epithelioid inflammatory myofibroblastic sarcoma with exceptionally long response to lorlatinib—a case report
Rafał Becht, Kajetan Kiełbowski, Justyna Żychowska, Wojciech Poncyljusz, Aleksandra Łanocha, Katarzyna Kozak, Ewa Gabrysz-Trybek, Paweł Domagała
Therapeutic Advances in Medical Oncology.2024;[Epub] CrossRef - Rare giant epithelioid inflammatory myofibroblastic sarcoma of the abdominal cavity in a child: a case report and review of the literature
Jinzhou Li, Haixing Su, Sheng Zhang, Xianyun Chen, Chongzhi Hou, Tao Cheng
Frontiers in Oncology.2024;[Epub] CrossRef - Case report: Epithelioid inflammatory myofibroblastic sarcoma treated with an ALK TKI ensartinib
Mengmeng Li, Ruyue Xing, Jiuyan Huang, Chao Shi, Chunhua Wei, Huijuan Wang
Frontiers in Oncology.2023;[Epub] CrossRef - Epithelioid Inflammatory Myofibroblastic Sarcoma With Poor Response to Crizotinib: A Case Report
Soheila Aminimoghaddam, Roghayeh Pourali
Clinical Medicine Insights: Case Reports.2023;[Epub] CrossRef - Epithelioid inflammatory myofibroblastic sarcoma: a case report and brief literature review
Weidong Dou, Yu Guan, Tao Liu, Hang Zheng, Shuo Feng, Yingchao Wu, Xin Wang, Zhanbing Liu
Frontiers in Oncology.2023;[Epub] CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
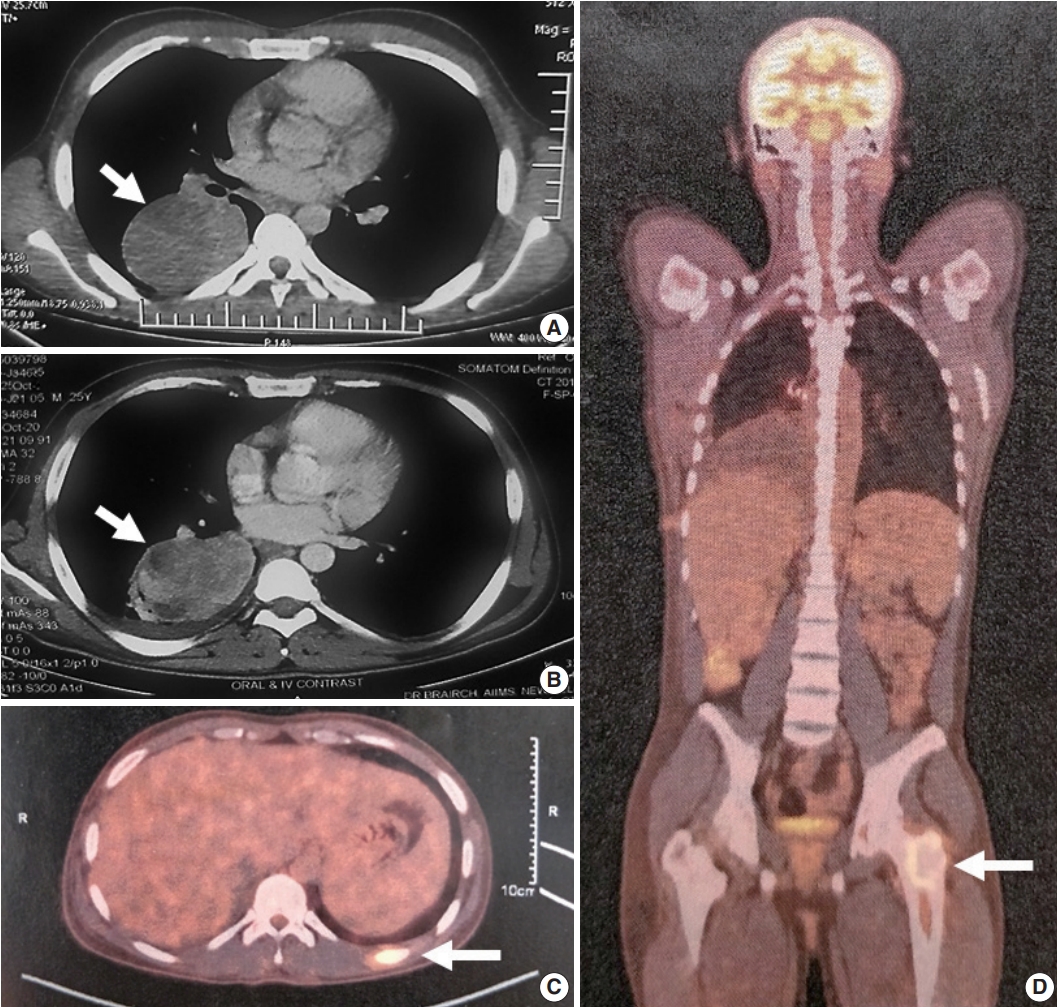
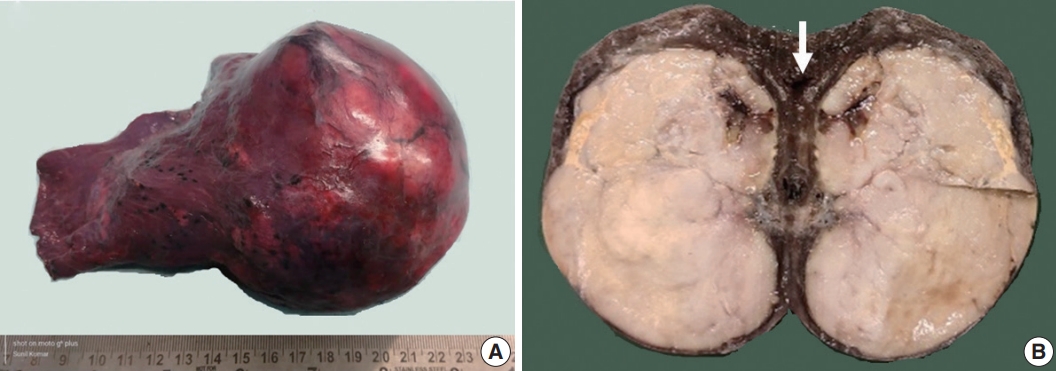
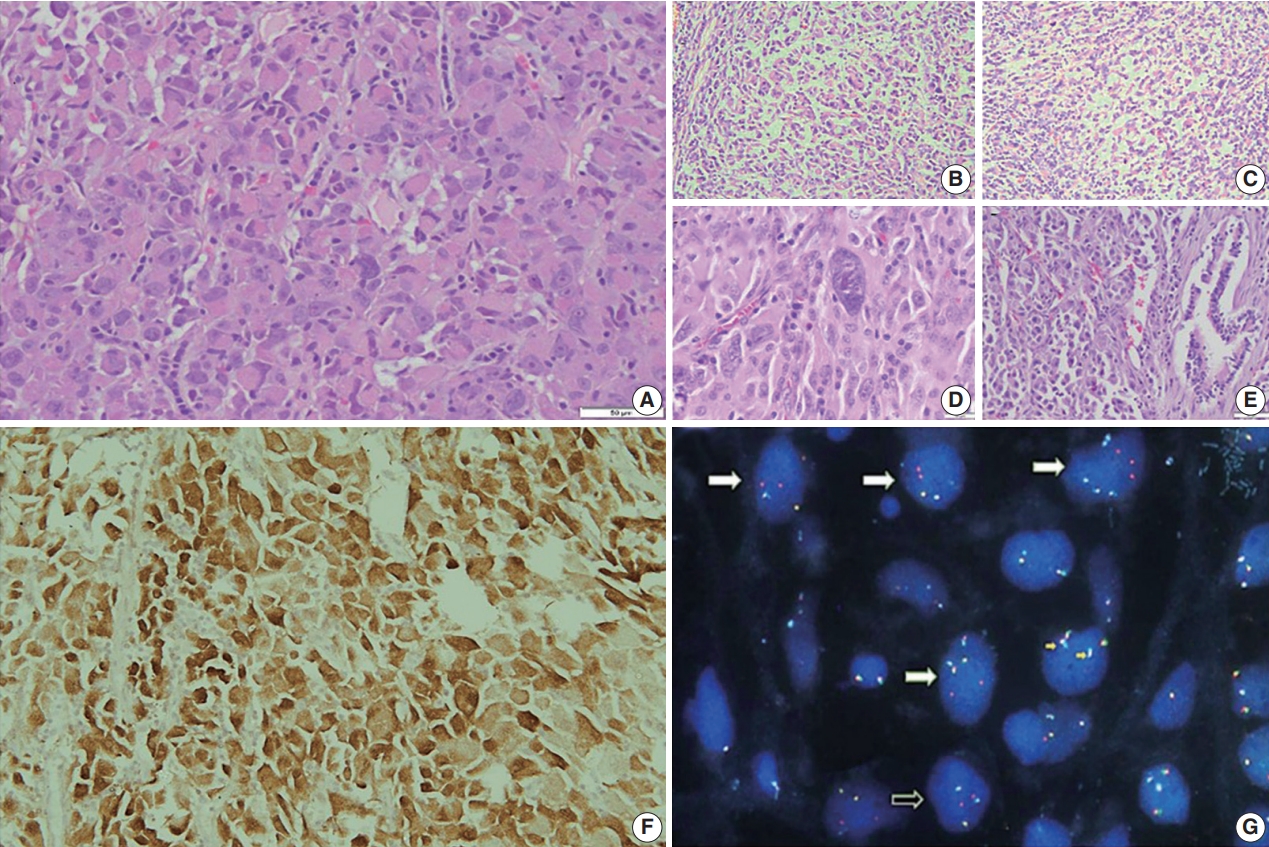



Fig. 1.
Fig. 2.
Fig. 3.
| No. | Study | Age (yr)/Sex | Anatomic site | Size (cm) | Clinical Presentation | Metastasis at presentation | Immunoprofile | Molecular confirmation (method) | Primary treatment | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Fu et al. [7] | 21/M | Left lower lobe lung | 10 | Weight loss, fatigue | None | Desmin+, ALK (c), CD30– | ALK rearrangement (FISH) | Lobectomy | Bone metastases (pelvis, vertebrae) and underwent laminectomy; death 4 mo after resection |
| 2 | Sarmiento et al. [8] | 71/M | Pleura based, left lower lobe | 12.5 | Dyspnoea, pleural effusion | None | NA | ALK rearrangement (FISH) | Lobectomy and adjuvant crizotinib | Progressed on crizotinib after 2 mo; near complete response to second line ALK inhibitor; remission after 1 yr at last follow-up |
| 3 | Kozu et al. [9] | 57/M | Pleural cavity/chest wall | NA | Pleural effusion and dyspnoea | N/A | Desmin+, cytokeratin+, ALK+ (c), CD30– | RANBP2-ALK fusion (PCR) | Crizotinib | NA |
| 4 | Present case | 25/M | Lung | 7 | Cough, loss of weight, fever, dyspnoea | Liver (solitary) | Desmin+, focal cytokeratin+, ALK+ (c), CD30– | ALK rearrangement (FISH) | Crizotinib | Complete resolution of liver metastases and stable pulmonary disease for 6 mo; bony metastases developed at 10 mo; lobectomy done and stable metastatic disease on crizotinib for 3 mo |
| Right lower lobe |
| Entity | Incidence as lung mass | Epithelial markers | Mesenchymal markers | Other markers | ALK protein expression | Commonest ALK fusions |
|---|---|---|---|---|---|---|
| IMT [2] | Common | Pan CK+/– | SMA+, Desmin+ | CD30– | Cytoplasmic (50%–60%) | TPM3-ALK, TPM4-ALK |
| ALCL [21] | Uncommon | EMA+ | SMA+, Desmin– | CD30+ | Nuclear and cytoplasmic | NPM-ALK |
| ALK+LBL [21] | Rare | EMA+ | – | CD138+, CD38+, Mum1/IRF1+, IgA+, Bob1+ | Cytoplasmic, nuclear and nucleolar | CLTC-ALK |
| ALK-SEC31A | ||||||
| ALK-rearranged NSCLC | Common | CK+ | – | Cytoplasmic | EML4-ALK [14] | |
| Melanoma | Extremely rare | CK– | – | S-100+, Melan A+, HMB45+, SOX10+ | ALKATI (seen in cutaneous melanoma) [22] | EML4-ALK [23] |
| Epithelioid mesothelioma [2] | Uncommon | CK5/6+/–, EMA+/– | Desmin– | Calretinin+/–, WT+, D2-40+ | – | – |
| Rhabdomyosarcoma | Rare | CK/EMA– | Desmin+, Actin+ | MyoD1+, Myogenin+ CD30– | Cytoplasmic | NPM-ALK [24], EML4-ALK [25] |
| Epithelioid leiomyosarcoma [1] | Rare | EMA+ | SMA+, Desmin+, h-caldesmon+ | CD34+/–CK+/–, EMA+/– | – | – |
| EIMS [2] | Rare | CK or EMA–/+ | Desmin+, SMA+ | CD30+ | Nuclear membrane or cytoplasmic with perinuclear accentuation | ALK-RRBP1 [3-5], ALK-RANBP2 |
| Pleomorphic carcinoma [2] | < 1% | NSCC component CK+, TTF-1 +, EMA+ | Vimentin+ | Surfactant protein A+, p53+ | Rare | – |
ALK, anaplastic lymphoma kinase; (c), cytoplasmic; FISH, fluorescent in situ hybridization; NA, not available; RANBP2, Ran specific binding protein 2; PCR, polymerase chain reaction.
ALK, anaplastic lymphoma kinase; ALK ATI, anaplastic lymphoma kinase with alternative transcription initiation; IMT, inflammatory myofibroblastic tumor; CK/PanCK, cytokeratin; SMA, smooth muscle actin; TPM, tropomyosin; ALCL, anaplastic large cell lymphoma; EMA, epithelial membrane antigen; NPM, nucleophosmin; Mum1/IRF1, multiple myeloma 1/interferon regulatory factor 4 protein; CLTC, clathrin heavy chain; NSCLC, non–small cell lung carcinoma; EML4, echinoderm microtubule-associated protein-like 4; HMB45, human melanoma black 45; EIMS, epithelioid Inflammatory myofibroblastic sarcoma;

