 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 58(5); 2024 > Article
-
Case Study
Rhabdomyosarcoma of the skull with EWSR1 fusion and ALK and cytokeratin expression: a case report -
Hyeong Rok An1
 , Kyung-Ja Cho1, Sang Woo Song2, Ji Eun Park3, Joon Seon Song1
, Kyung-Ja Cho1, Sang Woo Song2, Ji Eun Park3, Joon Seon Song1 -
Journal of Pathology and Translational Medicine 2024;58(5):255-260.
DOI: https://doi.org/10.4132/jptm.2024.08.15
Published online: September 5, 2024
1Department of Pathology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
2Department of Neurosurgery, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
3Department of Radiology and Research Institute of Radiology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
- Corresponding Author: Joon Seon Song, MD, PhD, Department of Pathology, Asan Medical Center, University of Ulsan College of Medicine, 88 Olympic-ro 43-gil, Songpa-gu, Seoul 05505, Korea Tel: +82-2-3010-4548, Fax: +82-2-472-7898, E-mail: songjs@amc.seoul.kr
© The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- Rhabdomyosarcoma (RMS) comprises of heterogeneous group of neoplasms that occasionally express epithelial markers on immunohistochemistry (IHC). We herein report the case of a patient who developed RMS of the skull with EWSR1 fusion and anaplastic lymphoma kinase (ALK) and cytokeratin expression as cytomorphologic features. A 40-year-old man presented with a mass in his forehead. Surgical resection was performed, during which intraoperative frozen specimens were obtained. Squash cytology showed scattered or clustered spindle and epithelioid cells. IHC revealed that the resected tumor cells were positive for desmin, MyoD1, cytokeratin AE1/AE3, and ALK. Although EWSR1 rearrangement was identified on fluorescence in situ hybridization, ALK, and TFCP2 rearrangement were not noted. Despite providing adjuvant chemoradiation therapy, the patient died of tumor progression 10 months after diagnosis. We emphasize that a subset of RMS can express cytokeratin and show characteristic histomorphology, implying the need for specific molecular examination.
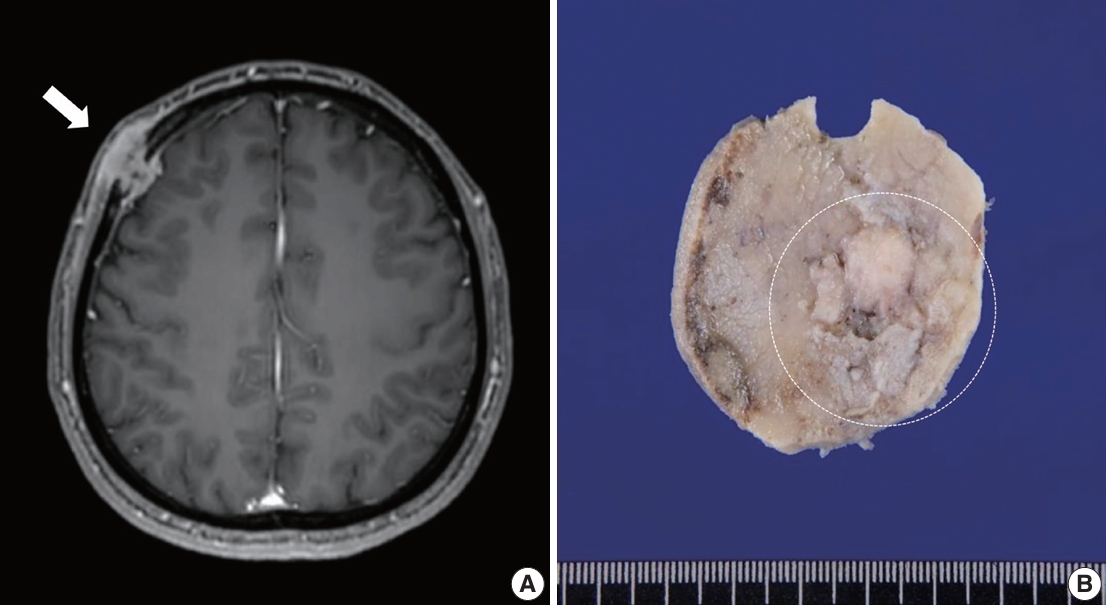
- A 40-year-old previously healthy man presented with a palpable, hard, unmovable mass in his right forehead that appeared 3 months prior. Magnetic resonance imaging detected a 2.7-cm-sized dumbbell-shaped enhancing mass in the right frontal bone involving the scalp and meninges (Fig. 1A). The mass was suspected to be a malignant bone tumor, such as Langerhans cell histiocytosis or metastasis.
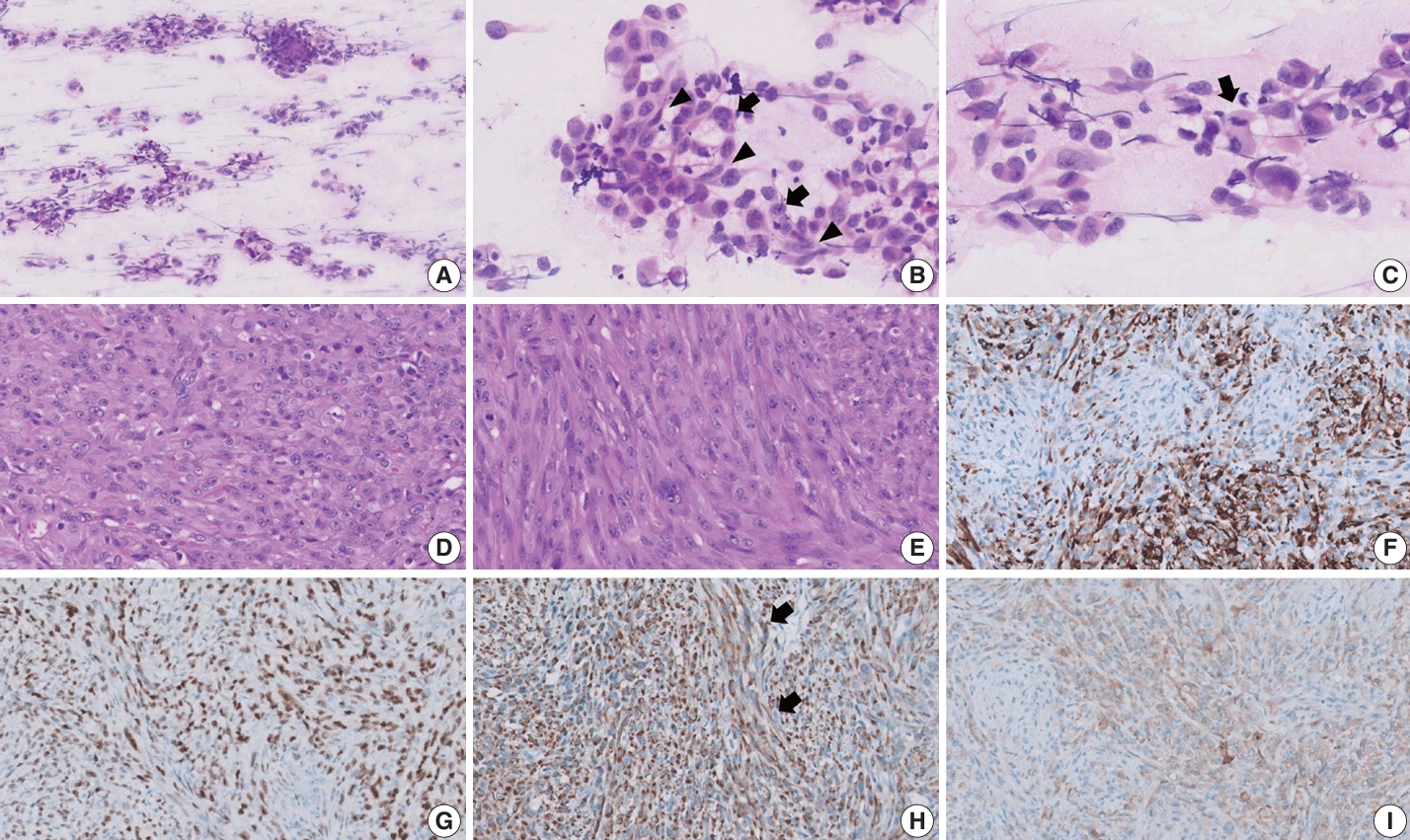
- Surgical resection was performed without biopsy, and samples from the scalp were obtained for intraoperative frozen section diagnosis. Squash cytology revealed small to large loosely cohesive clusters with smear artifact (Fig. 2A). The background was clear with few-to-no inflammatory cells and no necrosis. The clusters consisted of mixed epithelioid and spindle cells showing round-to-oval nuclei, prominent nucleoli, and granular chromatin with eosinophilic cytoplasm (Fig. 2B). Marked nuclear pleomorphism and scant mitotic activity were observed (Fig. 2C). Frozen section diagnosis confirmed positivity for malignancy.
- During gross examination, we observed an ill-demarcated, homogenous, whitish, solid, hard mass in the skull measuring 2.5×2.2×1.1 cm and penetrating through the dura mater (Fig. 1B). The resected tumor consisted of spindle and epithelioid cells with primarily a fascicular or whirling arrangement and few portions of solid growth (Fig. 2D, E). The neoplastic cells had round, vesicular nuclei, prominent one or two macronucleoli and moderate-to-abundant eosinophilic cytoplasm. Brisk mitosis and necrosis were noted.
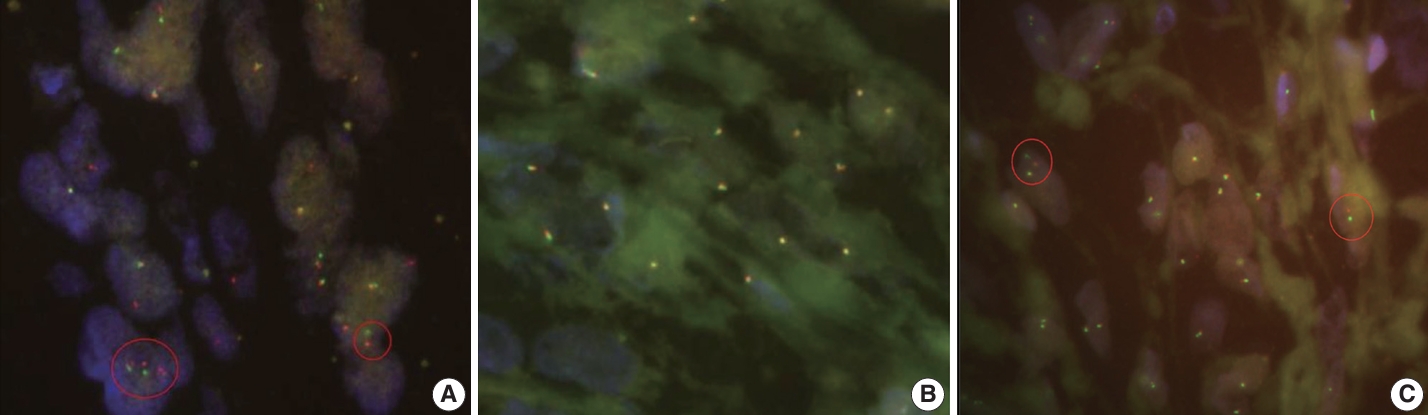
- The tumor cells were diffusely positive for desmin (clone D33, 1:200, Dako, Santa Clara, CA, USA), MyoD1 (EP212, 1:50, Cell Marque, Rocklin, CA, USA), and cytokeratin AE1/AE3 (1:400, Novocastra, San Jose, CA, USA) and focally positive for ALK (5A4, 1:200, Novocastra), smooth muscle actin (M0851, 1:500, Dako), myogenin (F5D, 1:100, Cell Marque), epithelial membrane antigen (EMA; E29, 1:100, Dako), p53 (DO-7, 1:1,000, Dako), S100 (4C4.9, 1:400, Cell Marque), nestin (10C2, 1:1,000, Cell Marque), SATB2 (polyclonal, 1:200, Cell Marque) on IHC (Fig. 2F–I). Conversely, the tumor cells tested negative for myoglobin (Z001, 1:1,000, Thermo Fisher Scientific, Waltham, MA, USA), signal transducer and activator of transcription 6 (polyclonal, 1:1,000, Abcam, Cambridge, UK), cyclin-dependent kinase 4 (DCS-31, 1:100, Santa Cruz Biotechnology, Santa Cruz, CA, USA), MDM-2 (SPM14, 1:50, Zeta, Sierra Madre, CA, USA), CD34 (1:400, Cell Marque), ERG (EP111, 1:400, Cell Marque), and HMB45 (1:50, Dako). EWSR1, ALK, and TFCP2 break-apart fluorescence in situ hybridization (FISH) revealed EWSR1 rearrangement (Fig. 3A) but no ALK or TFCP2 rearrangement (Fig. 3B, C). A molecular study using next-generation sequencing was attempted, but DNA quality was too degraded by decalcification.
- Although our patient received one cycle of adjuvant chemotherapy (vincristine, doxorubicin, and cyclophosphamide according to the IRS-III protocol), the size of the suspicious mass continued to increase for 2 months. No gross tumor remained at the time of surgery. Intensity-modulated radiation therapy (4680 cGY/26fx), three cycles of second-line chemotherapy (ifosfamide, carboplatin, and etoposide), and one cycle of third-line chemotherapy (gemcitabine and doxorubicin) were provided consecutively for residual/recurrent tumor. Unfortunately, the patient’s condition deteriorated with multiple metastases to the dura mater, skin, lung, rib, and abdominal cavity. The patient eventually died of tumor progression 10 months after diagnosis.
CASE REPORT
- RMSs occurring in the head and neck, including the parameningeal or orbital area, account for one-third of all cases of RMS in children and adolescents. Among adults, RMSs are most frequently observed in the extremities, followed by the chest/abdominal/pelvic, genitourinary, and head or neck regions [1]. However, RMSs of the skull are quite uncommon, with most cases originating from the skull base or temporal bone [8]. Primary tumors detected in the vault of the skull should be preferentially considered osteoma, osteosarcoma, Ewing sarcoma, Langerhans cell histiocytosis, plasmacytoma, or chondrosarcoma [9]. Moreover, for transdiploic lesions in the skull vault, radiologic differential diagnoses include meningioma, solitary fibrous tumor, lymphoma, plasmacytoma, and metastasis [10]. As the present case was initially suspected to be Langerhans cell histiocytosis or metastasis, establishing a correct diagnosis of RMS of the skull vault was challenging given its rarity in this location.
- The EWSR1 gene, which stands for Ewing sarcoma breakpoint region 1, is ubiquitously involved in various cellular processes, and its rearrangements with diverse partner genes have been associated with the development of multiple types of tumors [11]. For example, ESWR1 rearrangements have been frequently detected and serve as a key diagnostic finding in certain soft tissue tumors, such as Ewing sarcoma, round cell sarcomas with EWSR1–non-ETS fusions, desmoplastic small round cell tumor, myxoid liposarcoma, tumors with EWSR1/FUS fused to the CREB-family, sclerosing epithelioid fibrosarcoma, extraskeletal myxoid chondrosarcoma, and RMS with EWSR1/FUS-TFCP2 fusion. Considering the presence of EWSR1 fusion in various sarcomas, EWSR1-rearranged tumors necessitate further examination for the detection of the partner gene, especially in cases showing unusual clinicopathologic features. Despite having identified EWSR1 break-apart on FISH, we failed to confirm the counterpart gene to which the EWSR1 gene fused.
- The findings of the present report raise questions about a series of differential diagnoses, due to the present case’s unique and complex characteristics. EWSR1-PATZ1 and EWSR1-NFATC2 sarcomas are classified as round cell sarcomas with EWSR1–non-ETS fusions according to World Health Organization tumor classification and can show diverse histopathologic and immunophenotypic features [12]. EWSR1-PATZ1 sarcomas usually originate from the deep soft tissue of the chest wall and abdomen; however, they can develop in the head and neck as previously reported [13]. They express myogenic and neurogenic markers (S100P, SOX10, and GFAP) but rarely exhibit epithelial markers. Furthermore, ALK overexpression has not yet been reported in this tumor. Although EWSR1-NFATC2 sarcomas can show mixed epithelioid and spindle cell histology and dot-like positivity for cytokeratin, they are usually accompanied by abundant myxohyalinized stroma and do not express skeletal muscle differentiation [14]. A subset of tumors with EWSR1/FUS fused to the CREB-family has shown a predilection for intracranial location and exhibited both EMA and desmin [15]. These tumors consisted of ovoid or round cells in a myxoid background. Other EWSR1-CREB fusion neoplasms demonstrated a hybrid epithelioid and spindle morphology with cytokeratin and desmin positivity, and one expressed diffuse ALK positivity [16]. However, they commonly arise in mesothelial-lined cavities.
- RMSs with EWSR1/FUS-TFCP2 fusion are a newly emerging sub-classification characterized by a predilection for craniofacial bone, specific immunoprofiles, and molecular alterations [7,17]. This tumor comprises hybrid epithelioid and spindle cells. The latter are arranged in fascicular growth and have nuclei that are ovoid and fusiform with prominent nucleoli and mild pleomorphism, whereas the former contain an abundance of eosinophilic, often glassy cytoplasm arranged in solid sheets [18,19]. Several cases contain a portion of small round or rhabdoid cytologic features. Almost all cases have high mitotic rates and tumor necrosis. The tumor displayed positivity for myogenic markers, such as MyoD1, myogenin, and desmin. Interestingly, tumors in most cases show positivity for cytokeratin AE1/AE3 and ALK on IHC, and molecular studies often reveal ALK overexpression. One study showed that EWSR1/FUS-TFCP2 can activate ALK upregulation [20]. However, ALK rearrangement has not been reported previously [18]. Although we could not directly confirm TFCP2 rearrangement or fusion, we suspected that the tumor in the present case might have been RMS with EWSR1-TFCP2 fusion based on the tumor’s location in the craniofacial bone, the presence of mixed epithelioid and spindle cells, and the tumor’s immunopositivity for myogenic markers, cytokeratin, ALK, and EWSR1 rearrangement. According to a report on spindle cell RMS, aberrant keratin expression was a unique feature of RMS with EWSR1-TFCP2 fusion, which differed from spindle cell RMS harboring other fusions [6]. Additionally, a literature review revealed that EWSR1 rearrangement in RMS is exceedingly rare, and most of these cases involve RMS with EWSR1-TFCP2 fusion. However, some round cell sarcomas with EWSR1 fusion express skeletal muscle markers, which necessities differential diagnosis [21]. Meanwhile, we interpreted the results of TFCP2 FISH in our case as negative despite the identification of a few break-apart signals. Although Bin Xu et al. counted 200 nuclei and considered split signal of over 20% to be positive [18], currently no definite criteria for interpreting TFCP2 FISH results have been established.
- The prognosis of RMS with EWSR1/FUS-TFCP2 is incredibly poor, with a median survival time of less than 21 months (Table 1) [6,7,18,19]. Over half of patients develop local recurrence, regional lymph node metastasis, or distant metastasis to the bones and lungs [18]. Treatment options involve surgical resection, chemotherapy, and radiotherapy, all of which have limited efficacy. Although ALK inhibitors have been proposed to be a potential target therapy, their effectiveness remains unclear [20,22,23].
- In summary, we report an unusual case of RMS of the skull with EWSR1 fusion and ALK and cytokeratin expression, highlighting the importance of precise histopathologic examination along with comprehensive immunohistochemical and molecular evaluation for diagnosis and management.
DISCUSSION
Ethics Statement
This study was approved by the Institutional Review Board, and the need for informed consent was waived (Asan Medical Center IRB No. 2023-1548).
Availability of Data and Material
The datasets generated or analyzed during the study are available from the corresponding author on reasonable request.
Code Availability
Not applicable.
Author Contributions
Conceptualization: JSS. Data curation: HRA. Investigation: HRA, JSS. Resources: SWS, JEP, JSS. Supervision: JSS, KJC. Visualization: HRA, JSS. Writing—original draft preparation: HRA. Writing—review & editing: KJC, SWS, JEP, JSS. Approval of final manuscript: all authors.
Conflicts of Interest
J.S.S., a contributing editor of the Journal of Pathology and Translational Medicine, was not involved in the editorial evaluation or decision to publish this article. All remaining authors have declared no conflicts of interest.
Funding Statement
No funding to declare.
| Study | No. of cases | Age (yr) | Sex (M:F) | Locations | Outcomes |
|---|---|---|---|---|---|
| Le Loarer et al. (2020) [7] | 14 | 11–86 (mean, 31) | 3:4 | Craniofacial (8/14), other bone (4/14), soft tissue (2/12) | Median survival: 8 months |
| Chrisinger et al. (2020) [19] | 23a | 11–86 | 1:2.7 | Craniofacial (12/23), other bone (9/23), soft tissue (2/23) | Median survival: 15 months |
| Xu et al. (2021) [18] | 27a | 11–74 (mean, 25) | 1.25:1 | Craniofacial (18/27), other bone (8/27), soft tissue (1/27) | 1- and 2-year disease-specific survival rate: 74% and 35%, respectively |
| Dehner et al. (2023) [6] | 56a | 8–86 (mean, 34) | 5:9 | Craniofacial (37/56), other bone (13/56), soft tissue (6/56) | Median survival: 21 months |
- 1. Sultan I, Qaddoumi I, Yaser S, Rodriguez-Galindo C, Ferrari A. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: an analysis of 2,600 patients. J Clin Oncol 2009; 27: 3391-7. ArticlePubMed
- 2. Agaram NP, LaQuaglia MP, Alaggio R, et al. MYOD1-mutant spindle cell and sclerosing rhabdomyosarcoma: an aggressive subtype irrespective of age. A reappraisal for molecular classification and risk stratification. Mod Pathol 2019; 32: 27-36. ArticlePubMedPMCPDF
- 3. Mosquera JM, Sboner A, Zhang L, et al. Recurrent NCOA2 gene rearrangements in congenital/infantile spindle cell rhabdomyosarcoma. Genes Chromosomes Cancer 2013; 52: 538-50. ArticlePubMedPMC
- 4. Alaggio R, Zhang L, Sung YS, et al. A molecular study of pediatric spindle and sclerosing rhabdomyosarcoma: identification of novel and recurrent VGLL2-related fusions in infantile cases. Am J Surg Pathol 2016; 40: 224-35. PubMedPMC
- 5. Bahrami A, Gown AM, Baird GS, Hicks MJ, Folpe AL. Aberrant expression of epithelial and neuroendocrine markers in alveolar rhabdomyosarcoma: a potentially serious diagnostic pitfall. Mod Pathol 2008; 21: 795-806. ArticlePubMedPDF
- 6. Dehner CA, Broski SM, Meis JM, et al. Fusion-driven spindle cell rhabdomyosarcomas of bone and soft tissue: a clinicopathologic and molecular genetic study of 25 cases. Mod Pathol 2023; 36: 100271.ArticlePubMed
- 7. Le Loarer F, Cleven AH, Bouvier C, et al. A subset of epithelioid and spindle cell rhabdomyosarcomas is associated with TFCP2 fusions and common ALK upregulation. Mod Pathol 2020; 33: 404-19. ArticlePubMedPDF
- 8. Radzikowska J, Kukwa W, Kukwa A, Czarnecka A, Krzeski A. Rhabdomyosarcoma of the head and neck in children. Contemp Oncol (Pozn) 2015; 19: 98-107. PubMedPMC
- 9. Kakkar A, Nambirajan A, Suri V, et al. Primary bone tumors of the skull: spectrum of 125 cases, with review of literature. J Neurol Surg B Skull Base 2016; 77: 319-25. ArticlePubMedPMC
- 10. Pons Escoda A, Naval Baudin P, Mora P, et al. Imaging of skull vault tumors in adults. Insights Imaging 2020; 11: 23.PubMedPMC
- 11. Flucke U, van Noesel MM, Siozopoulou V, et al. EWSR1: the most common rearranged gene in soft tissue lesions, which also occurs in different bone lesions: an updated review. Diagnostics (Basel) 2021; 11: 1093.ArticlePubMedPMC
- 12. WHO Classification of Tumours Editorial Board. WHO classification of tumours series, 5th ed. Vol. 3. Soft tissue and bone tumours. Lyon: International Agency for Research on Cancer, 2020.
- 13. Bridge JA, Sumegi J, Druta M, et al. Clinical, pathological, and genomic features of EWSR1-PATZ1 fusion sarcoma. Mod Pathol 2019; 32: 1593-604. ArticlePubMedPDF
- 14. Wang GY, Thomas DG, Davis JL, et al. EWSR1-NFATC2 translocation-associated sarcoma clinicopathologic findings in a rare aggressive primary bone or soft tissue tumor. Am J Surg Pathol 2019; 43: 1112-22. ArticlePubMed
- 15. Kao YC, Sung YS, Zhang L, et al. EWSR1 fusions with CREB family transcription factors define a novel myxoid mesenchymal tumor with predilection for intracranial location. Am J Surg Pathol 2017; 41: 482-90. ArticlePubMedPMC
- 16. Argani P, Harvey I, Nielsen GP, et al. EWSR1/FUS-CREB fusions define a distinctive malignant epithelioid neoplasm with predilection for mesothelial-lined cavities. Mod Pathol 2020; 33: 2233-43. ArticlePubMedPMCPDF
- 17. Watson S, Perrin V, Guillemot D, et al. Transcriptomic definition of molecular subgroups of small round cell sarcomas. J Pathol 2018; 245: 29-40. ArticlePubMedPDF
- 18. Xu B, Suurmeijer AJ, Agaram NP, Zhang L, Antonescu CR. Head and neck rhabdomyosarcoma with TFCP2 fusions and ALK overexpression: a clinicopathological and molecular analysis of 11 cases. Histopathology 2021; 79: 347-57. ArticlePubMedPMCPDF
- 19. Chrisinger JSA, Wehrli B, Dickson BC, et al. Epithelioid and spindle cell rhabdomyosarcoma with FUS-TFCP2 or EWSR1-TFCP2 fusion: report of two cases. Virchows Arch 2020; 477: 725-32. ArticlePubMedPDF
- 20. Schopf J, Uhrig S, Heilig CE, et al. Multi-omic and functional analysis for classification and treatment of sarcomas with FUS-TFCP2 or EWSR1-TFCP2 fusions. Nat Commun 2024; 15: 51.PubMedPMC
- 21. Michal M, Rubin BP, Agaimy A, et al. EWSR1-PATZ1-rearranged sarcoma: a report of nine cases of spindle and round cell neoplasms with predilection for thoracoabdominal soft tissues and frequent expression of neural and skeletal muscle markers. Mod Pathol 2021; 34: 770-85. ArticlePubMedPDF
- 22. Brunac AC, Laprie A, Castex MP, et al. The combination of radiotherapy and ALK inhibitors is effective in the treatment of intraosseous rhabdomyosarcoma with FUS-TFCP2 fusion transcript. Pediatr Blood Cancer 2020; 67: e28185. ArticlePubMedPDF
- 23. Valerio E, Furtado Costa JL, Perez Fraile NM, et al. Intraosseous spindle cell/epithelioid rhabdomyosarcoma with TFCP2 rearrangement: a recent recognized subtype with partial response to alectinib. Int J Surg Pathol 2023; 31: 861-5. PubMed
REFERENCES
Figure & Data
References
Citations
- Rhabdomyosarcomas of Bone
Ahmed Shah, Andrew L. Folpe
Surgical Pathology Clinics.2025; 18(3): 503. CrossRef - Review of imaging modalities and radiological findings of calvarial lesions
Erkan Gökçe, Murat Beyhan
World Journal of Radiology.2025;[Epub] CrossRef - Molecular Morphology of Telangiectatic Osteosarcoma Associated With Сystic Content: A Case Report
David Makaridze, Armaz Mariamidze, Tamuna Gvianishvili, Giulia Ottaviani , Liana Gogiashvili
Cureus.2025;[Epub] CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
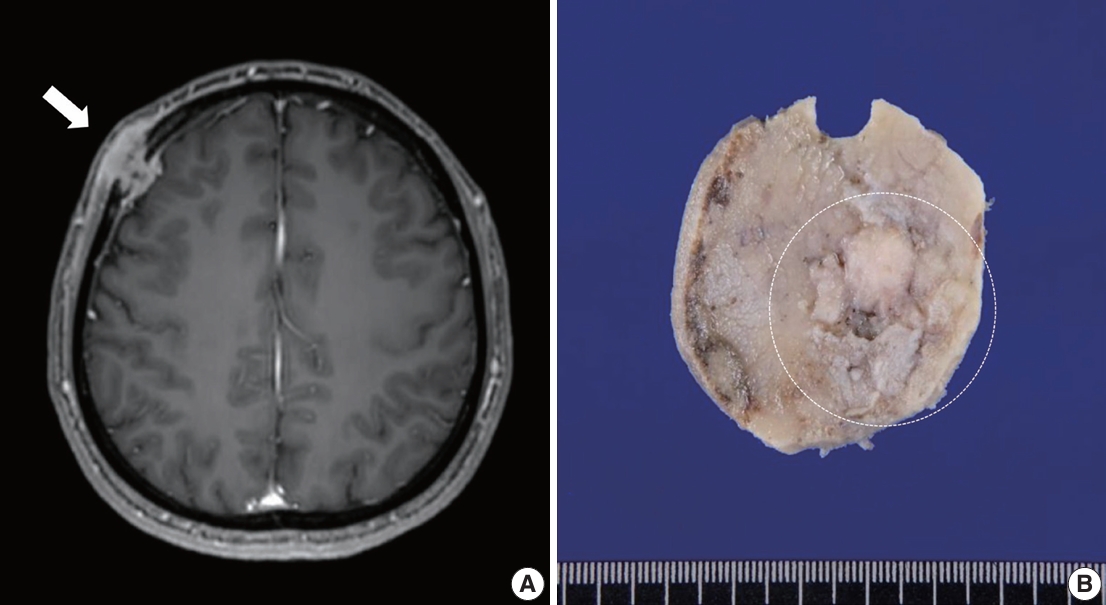
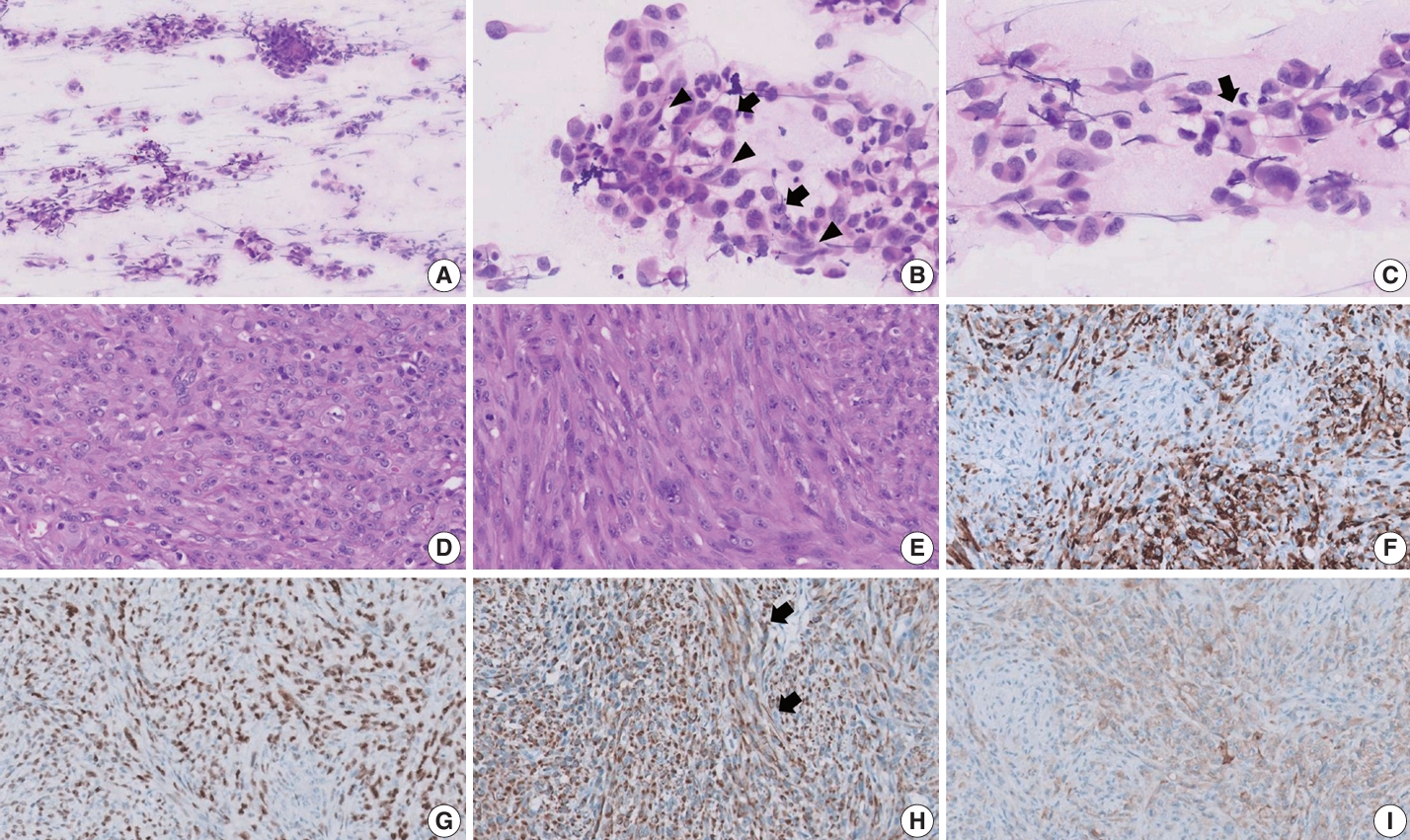
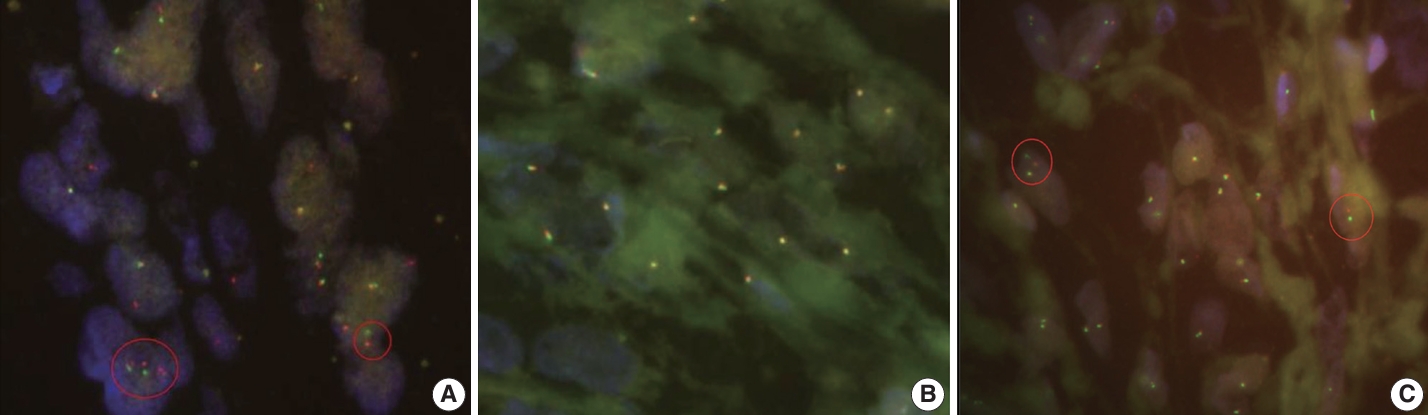

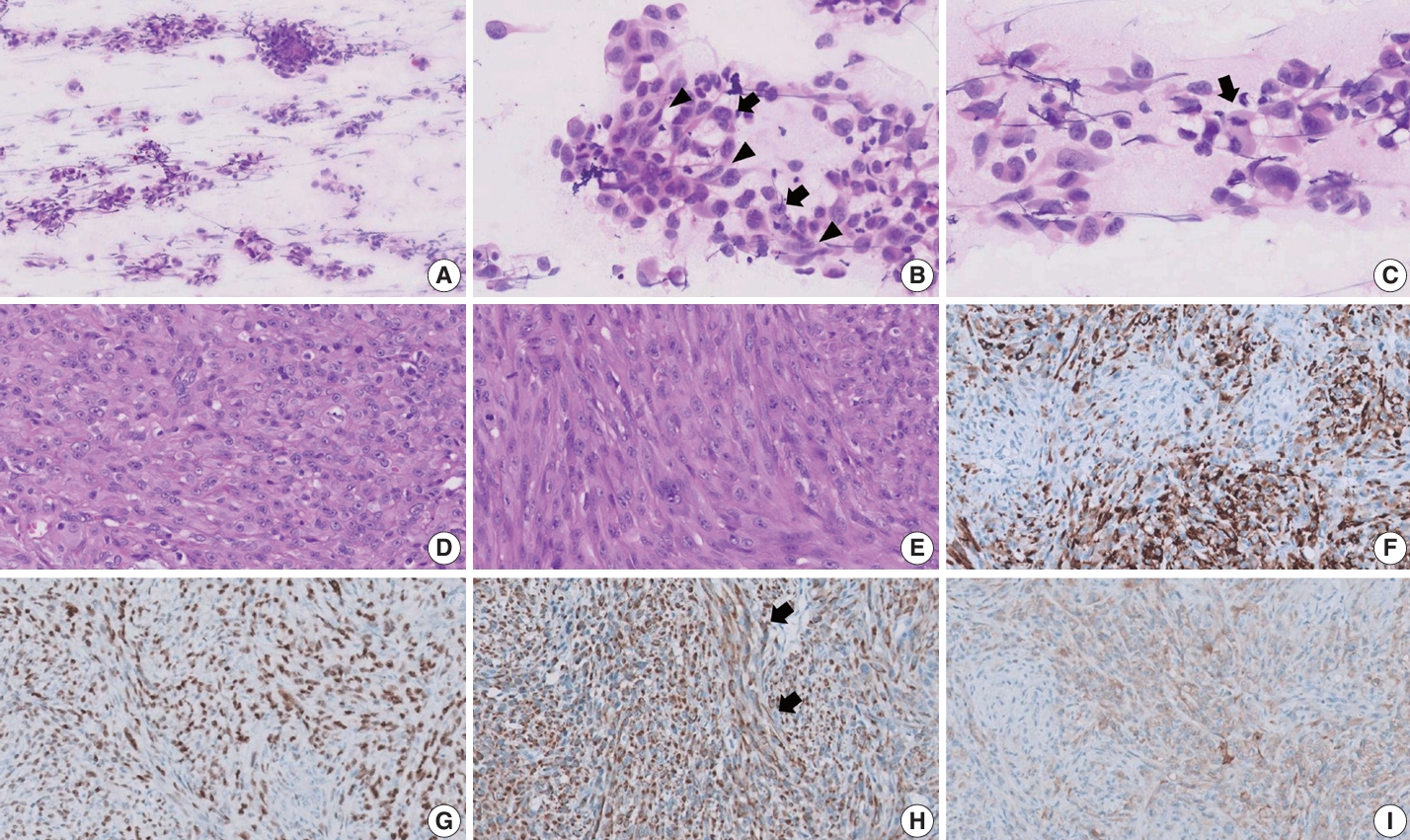
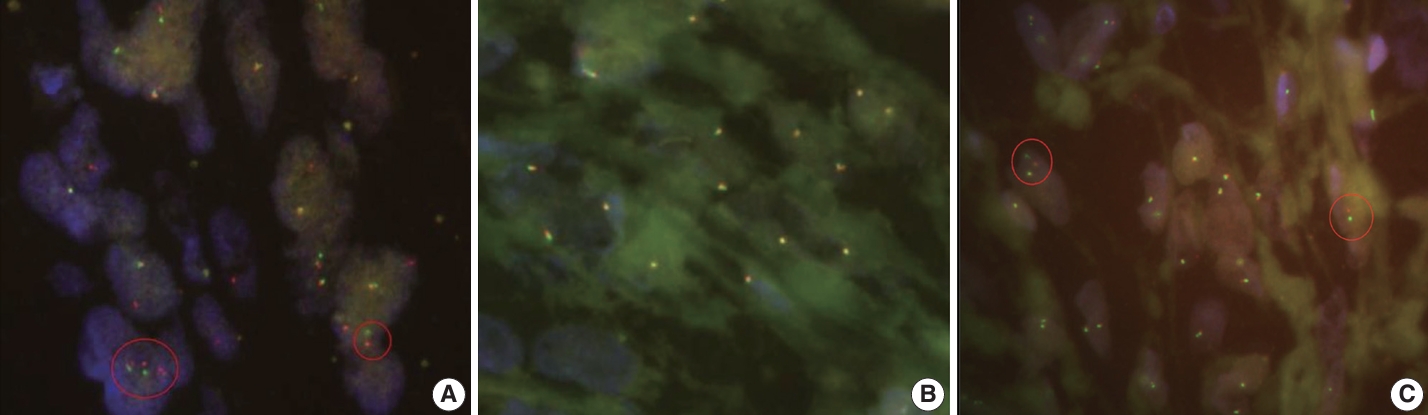
Fig. 1.
Fig. 2.
Fig. 3.
| Study | No. of cases | Age (yr) | Sex (M:F) | Locations | Outcomes |
|---|---|---|---|---|---|
| Le Loarer et al. (2020) [7] | 14 | 11–86 (mean, 31) | 3:4 | Craniofacial (8/14), other bone (4/14), soft tissue (2/12) | Median survival: 8 months |
| Chrisinger et al. (2020) [19] | 23 |
11–86 | 1:2.7 | Craniofacial (12/23), other bone (9/23), soft tissue (2/23) | Median survival: 15 months |
| Xu et al. (2021) [18] | 27 |
11–74 (mean, 25) | 1.25:1 | Craniofacial (18/27), other bone (8/27), soft tissue (1/27) | 1- and 2-year disease-specific survival rate: 74% and 35%, respectively |
| Dehner et al. (2023) [6] | 56 |
8–86 (mean, 34) | 5:9 | Craniofacial (37/56), other bone (13/56), soft tissue (6/56) | Median survival: 21 months |
This number includes data from previously reported cases through a literature review.

