 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 58(6); 2024 > Article
-
Newsletter
What’s new in neuropathology 2024: CNS WHO 5th edition updates -
Heather Smith
 , Jared T. Ahrendsen
, Jared T. Ahrendsen -
Journal of Pathology and Translational Medicine 2024;58(6):346-349.
DOI: https://doi.org/10.4132/jptm.2024.09.11
Published online: September 30, 2024
Department of Pathology, Northwestern University Feinberg School of Medicine, Chicago, IL, USA
- Corresponding Author: Jared T. Ahrendsen, MD, PhD Department of Pathology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA E-mail: jared.ahrendsen@nm.org
This article has been published jointly, with consent, in both Journal of Pathology and Translational Medicine and PathologyOutlines.com.
• Received: August 17, 2024 • Accepted: September 11, 2024
© The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- Abstract
- GENERAL CHANGES IN TERMINOLOGY & TAXONOMY
- ADULT-TYPE DIFFUSE GLIOMAS
- PEDIATRIC-TYPE DIFFUSE LOW-GRADE GLIOMAS
- PEDIATRIC-TYPE DIFFUSE HIGH-GRADE GLIOMAS
- CIRCUMSCRIBED ASTROCYTIC GLIOMAS
- GLIONEURONAL AND NEURONAL TUMORS
- EPENDYMAL TUMORS
- EMBRYONAL TUMORS
- MENINGIOMAS
- MESENCHYMAL AND PERIPHERAL NERVE SHEATH TUMORS
- PITUITARY AND PINEAL TUMORS
- Meet the Authors
Abstract
- The fifth edition of the World Health Organization (WHO) Classification of Central Nervous System (CNS) Tumors was released in 2021, just five years following the updated fourth edition. Advanced molecular testing such as next-generation sequencing, RNA fusion analysis, and DNA methylation profiling has led to more precise grading and classification of pre-existing tumor types as well as the recognition of new ones. Herein, we outline the major updates of the 2021 WHO Classification of CNS tumors, with emphasis on the expanded molecular characterization of CNS tumors.
- • The words “entity” and “variant” have been replaced by “type” and “subtype.” For example, glioblastoma, isocitrate dehydrogenase (IDH)-wildtype is a type of tumor, and its many morphologic variants are now listed as subtypes.
- • CNS WHO grades, previously listed in Roman numerals (I, II, III, IV), are now listed in Arabic numerals (1, 2, 3, 4).
- • Integrated diagnoses combining both histologic and molecular features (e.g., astrocytoma, IDH-mutant) are now recommended.
- • Layered reporting to include integrated diagnosis, histopathological classification, CNS WHO grade, and relevant molecular information is recommended.
- • Grading is now done within tumor types rather than across types:
- ° E.g., astrocytoma, IDH-mutant is assigned a CNS grade of either 2, 3, or 4.
- ° Modifier terms such as “anaplastic” are no longer routinely included (e.g., the term “anaplastic astrocytoma” is no longer recommended).
- • Molecular characteristics are incorporated into the classification and grading of more tumors than in previous editions.
- • The Human Genome Organisation (HUGO) Gene Nomenclature Classification System is used for gene symbols and names, the Human Genome Variation Society recommendations for gene variants, and the International System for Cytogenetic Nomenclature recommendations for copy number variants.
- • “Not otherwise specified (NOS)” and “Not elsewhere classified (NEC)” are now included as diagnostic labels:
- ° “Not otherwise specified (NOS)” is applied when the molecular testing required for a specific diagnosis cannot be performed or fails to yield results.
- ° “Not elsewhere classified (NEC)” is applied when a comprehensive molecular workup is performed and the tumor does not conform to a recognized WHO tumor type.
- • Nineteen new and three provisional tumor types are included, described below.
GENERAL CHANGES IN TERMINOLOGY & TAXONOMY
- • Adult-type diffuse gliomas are now classified as one of three types based on the presence/absence of IDH mutation and 1p/19q-codeletion and graded within each of these types.
- ° “Oligoastrocytoma,” previously defined as a tumor with morphologic features of both oligodendroglioma and astrocytoma, is no longer recognized as a distinct entity.
- ° The “anaplastic” modifier is no longer applied to grade 3 tumors (e.g., the term “anaplastic oligodendroglioma” is no longer recommended).
- ° Because of fundamental differences in natural history and tumor biology, IDH-mutant gliomas are no longer called “glioblastoma” even when a CNS WHO grade of 4 can be assigned; the term “glioblastoma” is reserved specifically for IDH-wildtype adult-type diffuse gliomas.
- • Glioblastoma, IDH-wildtype:
- ° Absence of IDH and histone H3 mutations;
- ° Presence of either microvascular proliferation or tumor cell necrosis;
- ° If lacking the above histologic features, the presence of a TERT promoter mutation, EGFR amplification, or concurrent gain of chromosome 7 and loss of chromosome 10 (+7/-10) are sufficient for diagnosis.
- • Astrocytoma, IDH-mutant:
- ° Presence of mutation in IDH1 or IDH2;
- ° Absence of 1p/19q-codeletion;
- ° CNS WHO grade 2-4 assigned based on histologic features (as described in previous classifications), with the addition of CDKN2A/B homozygous deletion as a molecular criterion for grade 4 designation.
- • Oligodendroglioma, IDH-mutant and 1p/19q-codeleted:
- ° Presence of IDH1 or IDH2 mutation and concurrent 1p/19q-codeletion required for diagnosis;
- ° CNS WHO grade 2-3, with same histology-based criteria as previous classification scheme.
- • “Adult-type” and “pediatric-type” gliomas have been separated due to distinct molecular characteristics, biological behavior, and predilection for certain age groups. However, adult-type tumors can occasionally affect children, and pediatric-type gliomas are sometimes encountered in adult patients.
ADULT-TYPE DIFFUSE GLIOMAS
- • Each tumor type demonstrates diffuse growth pattern without high-grade morphologic features.
- • Diffuse astrocytoma, MYB- or MYBL1-altered (new entity): bland, monomorphic cells; characterized by fusion events involving MYB or MYBL1 (CNS WHO grade 1).
- • Angiocentric glioma: also defined by MYB alterations; however, it displays prominent perivascular growth and characteristically harbors the MYB::QKI fusion (CNS WHO grade 1).
- • Polymorphous low-grade neuroepithelial tumor of the young (PLNTY, new entity): CNS WHO grade 1 epileptogenic tumor that tends to have oligodendrocyte-like tumor cells, strong CD34 expression, calcifications (Fig. 1), and MAPK pathway alterations (most commonly BRAF p.V600E or fusions involving FGFR2/3).
- • Diffuse low-grade glioma, MAPK pathway-altered (new entity): diffuse growth and bland low-grade histomorphology; molecularly defined by the absence of mutations in IDH and histone H3, absence of homozygous CDKN2A/B deletion, and the presence of a MAPK pathway alteration (most commonly BRAF p.V600E or internal tandem duplication in the kinase domain of FGFR1). Specific CNS WHO grade not currently assigned.
PEDIATRIC-TYPE DIFFUSE LOW-GRADE GLIOMAS
- • Each tumor type demonstrates diffusely infiltrative growth, high-grade morphologic features, and variably aggressive clinical behavior.
- • Diffuse midline glioma, H3 K27-altered: CNS WHO grade 4 diffusely infiltrating glioma involving a midline structure (e.g., brainstem, thalamus, spinal cord), loss of H3K27me3 expression, and one of the following molecular alterations:
- ° Mutation in one of the genes encoding histone H3, leading to the substitution of lysine with either methionine or isoleucine at position 27 (K27M/I);
- ° An activating mutation or amplification of EGFR;
- ° EZHIP overexpression.
- • Diffuse hemispheric glioma, H3 G34-mutant (new entity): CNS WHO grade 4 diffusely infiltrating glioma involving the cerebral hemispheres, often with loss of nuclear ATRX expression; defined by a mutation in the histone H3-3A gene leading to the substitution of glycine with either arginine or valine at position 34 (G34R/V).
- • Diffuse pediatric-type high-grade glioma, H3- and IDH-wildtype (new entity): CNS WHO grade 4 glioma with high-grade histologic features divided into three subtypes based on DNA methylation profiling and enrichment for particular molecular alterations:
- ° pHGG RTK I: frequent PDGFRA amplification, intermediate prognosis (median overall survival of 21 months), frequently associated with prior radiation therapy or constitutional mismatch repair deficiency;
- ° pHGG RTK II: frequent TERT promoter mutation and EGFR amplification (similar to glioblastoma, IDH-wildtype), tends to have better prognosis (median overall survival of 44 months);
- ° pHGG MYCN: frequent MYCN amplification, poor prognosis (median overall survival of 14 months).
- • Infant-type hemispheric glioma (new entity): diffusely infiltrating glioma involving the cerebral hemispheres that tends to occur during the first year of life and is driven by fusion events involving NTRK1/2/3, ALK, MET, or ROS1. Specific CNS WHO grade not currently assigned.
PEDIATRIC-TYPE DIFFUSE HIGH-GRADE GLIOMAS
- • High-grade astrocytoma with piloid features (HGAP, new entity): newly recognized tumor type and one of the first to be defined by a specific DNA methylation profile, rather than by histomorphology or specific molecular alteration. The majority have morphologic features reminiscent of pilocytic astrocytoma or pleomorphic xanthoastrocytoma, molecular alterations involving the MAPK pathway, homozygous deletion of CDKN2A/B, and mutation or lost expression of ATRX. Specific CNS WHO grade not currently assigned.
- • Astroblastoma, MN1-altered: tumor cells arranged in perivascular pseudorosettes with reverse nuclear polarity and now defined genetic alteration (usually fusion event) involving MN1.
- • Chordoid glioma: chordoid morphology with recurrent PRKCA p.D463H mutation and characteristically located in the third ventricle (though no longer termed “chordoid glioma of the third ventricle”).
CIRCUMSCRIBED ASTROCYTIC GLIOMAS
- • Two new and one provisional tumor types, including:
- ° Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters (DGONC, provisional entity): variably differentiated cells with oligodendroglioma-like perinuclear halos, scattered multinucleated cells, and nuclear clusters. Molecular findings include monosomy 14 and a distinct DNA methylation profile. Specific CNS WHO grade not currently assigned.
- ° Myxoid glioneuronal tumor (new entity): CNS WHO grade 1 neoplasm composed of oligodendrocyte-like cells in a myxoid stroma (Fig. 2), often with floating neurons and neurocytic rosettes. Typically arises in the septal nuclei, periventricular white matter, or corpus callosum. Recurrent PDGFRA p.K385L/I mutation.
- ° Multinodular and vacuolating neuronal tumor (new entity): monomorphic neuronal elements arranged in clusters with vacuolated cells and matrix (CNS WHO grade 1). These tend to have MAPK pathway activating mutations, most commonly in MAP2K1 or BRAF (not p.V600E).
GLIONEURONAL AND NEURONAL TUMORS
- • Ependymal tumors are now classified by a combination of tumor location, histopathology, and molecular features. Ependymoma tumor types form distinct molecular-anatomic clusters that can be identified by DNA methylome analysis.
- • “Anaplastic ependymoma” is no longer a listed entity.
- • Supratentorial, posterior fossa, and spinal ependymomas can be assigned a CNS WHO grade of 2 or 3 based on the presence of microvascular proliferation, brisk mitotic activity, and necrosis (similar to prior classification schemes).
- • Supratentorial ependymomas:
- ° Supratentorial ependymoma, ZFTA fusion-positive: The majority have fusions involving ZFTA (formerly C11orf95) with RELA.
- ° Supratentorial ependymoma, YAP1 fusion-positive (new entity): The most common fusion partner is MAMLD1.
- • Posterior fossa ependymomas are divided into two methylation classes:
- ° Posterior fossa group A (PFA, new entity): defined by loss of H3K27me3 expression. These occur primarily in children and are locally aggressive.
- ° Posterior fossa group B (PFB, new entity): defined by retained H3K27me3 expression. These are more common in adults and show more indolent behavior.
- • Spinal ependymomas have their own methylation cluster and tend to show 22q loss and NF2 mutations.
- ° Spinal ependymoma, MYCN amplified (new entity): high-grade features are common, including anaplasia, elevated mitotic activity, microvascular proliferation, and necrosis. These are clinically more aggressive than conventional spinal ependymomas.
- • Myxopapillary ependymomas are now assigned a CNS WHO grade of 2 instead of 1 due to their tendency to recur and disseminate.
EPENDYMAL TUMORS
- • Medulloblastomas are now classified by a combination of molecular and histologic parameters. Two new and one provisional embryonal tumor types are also now described.
- • Medulloblastomas are still divided into four molecular groups, and all are assigned a CNS WHO grade 4:
- ° Medulloblastoma, WNT-activated;
- ° Medulloblastoma, SHH-activated and TP53-mutant;
- ° Medulloblastoma, SHH-activated and TP53-wildtype;
- ° Medulloblastoma, non-WNT/non-SHH.
- • Histopathologic classification of medulloblastoma (classic, desmoplastic/nodular, medulloblastoma with extensive nodularity, and large cell/anaplastic) are now considered morphologic patterns and summarized in one section.
- • Embryonal tumor with multilayered rosettes (ETMR): defined as having either a C19MC alteration or DICER1 mutation and includes the previously described embryonal tumor with abundant neuropil and true rosettes (ETANTR), medulloepithelioma, and ependymoblastoma.
- • Atypical teratoid/rhabdoid tumor (AT/RT) is now required to have SMARCB1 or SMARCA4 loss (or DNA methylation profile aligned with AT/RT).
- • Two new and one provisional tumor types:
- ° Cribriform neuroepithelial tumor (provisional entity): neuroepithelial tumor with cribriform architecture, periventricular location, and SMARCB1 loss.
- ° CNS neuroblastoma, FOXR2-activated (new entity): defined by FOXR2 rearrangements. These show varying amounts of neuroblastic and neuronal differentiation, and their cell of origin remains unknown. Assigned CNS WHO grade 4.
- ° CNS tumor with BCOR internal tandem duplication (new entity): CNS embryonal tumor (CNS WHO grade 4) defined by an internal duplication in exon 15 of BCOR.
EMBRYONAL TUMORS
- • Now considered one tumor type with multiple morphologic subtypes
- • Terms “atypical meningioma” and “anaplastic meningioma” are retained, unlike other tumor types described above.
- • Morphologic assignment of CNS WHO grades 1-3 is generally unchanged from prior edition.
- • New molecular criteria for meningioma grading: the presence of TERT promoter mutation or homozygous CDKN2A/B deletion now warrants designation as CNS WHO grade 3.
MENINGIOMAS
- • Nomenclature changes include the following:
- ° Hemangiopericytoma is now called solitary fibrous tumor, similar to the nomenclature change in the WHO Classification of Bone & Soft tissues, though grading criteria differ between CNS and extra-CNS tumors;
- ° Melanotic schwannoma is now called malignant melanotic nerve sheath tumor;
- ° Mesenchymal chondrosarcoma is now listed as a distinct tumor type;
- ° Paraganglioma is now referred to as cauda equina neuroendocrine tumor.
- • New and provisional tumor types include the following:
- ° Intracranial mesenchymal tumor, FET::CREB fusion-positive (provisional entity): mesenchymal tumor with widely variable architecture ranging from sheet-like to cord-like but generally has a collagenous or myxoid stroma, relatively low mitotic activity, and hemangioma-like collections of dilated, thin-walled vessels. Defined by a fusion involving a member of the FET RNA-binding protein family (most commonly EWSR1) with a member of the CREB family of transcription factors (CREB1, ATF1, or CREM).
- ° CIC-rearranged sarcoma (new entity): highgrade, poorly differentiated sarcoma (similar to those arising outside of the CNS) and defined by a fusion involving CIC (most often NUTM1).
- ° Primary intracranial sarcoma, DICER1-mutant (new entity): defined by mutations in DICER1 (somatic or germline) and usually consists of spindled to pleomorphic cells, characteristic cytoplasmic eosinophilic globules, and abundant mitotic activity (Fig. 3).
MESENCHYMAL AND PERIPHERAL NERVE SHEATH TUMORS
- • The following are changes in pituitary tumor nomenclature:
- ° Adamantinomatous craniopharyngioma and papillary craniopharyngioma, which have distinct morphology and molecular drivers, are now considered distinct tumor types.
- ° Pituitary adenoma is now called pituitary neuroendocrine tumor (PitNET).
- • New tumor types include the following:
- ° Pituitary blastoma (new entity): usually driven by alterations in DICER1 and composed of large anterior pituitary neuroendocrine cells, cuboidal or columnar primitive Rathke pouch epithelium, and small undifferentiated blastemal cells.
- ° Desmoplastic myxoid tumor of the pineal region, SMARCB1-mutant (new entity): extremely rare tumor with relatively bland spindled to epithelial cells arranged in cords within myxoid and heavily collagenized stroma. These are driven by alterations in SMARCB1 (Fig. 4).
PITUITARY AND PINEAL TUMORS
- Dr. Heather Smith is new to PathologyOutlines.com, starting in 2024. She completed her pathology residency at the University of Chicago and is currently a neuropathology fellow at Northwestern University Feinberg School of Medicine in Chicago, IL. Her clinical and research interests include surgical neuropathology, molecular classification of brain tumors, and bone/soft tissue pathology.
- Dr. Jared T. Ahrendsen joined the Department of Pathology as an Assistant Professor of Pathology at Northwestern University Feinberg School of Medicine in 2022, where he specializes in neuropathology and autopsy pathology. Dr. Ahrendsen is also a trained forensic pathologist and does forensic neuropathology consultations. He has been writing for PathologyOutlines.com since 2021 and joined the neuropathology editorial board in 2023. He received the PathologyOutlines.com Best Author Award in 2022 and 2023. Dr. Ahrendsen’s research interests include molecular classification of brain tumors and the utilization of post-mortem brain tissue to better understand human neurologic disease and injury.
Meet the Authors
Fig. 1.Polymorphous low-grade neuroepithelial tumor of the young (PLNTY). Low-grade neoplasm demonstrating diffuse growth pattern and microcalcifications (top panel). Strong, regional CD34 expression by tumor cells (bottom panel).
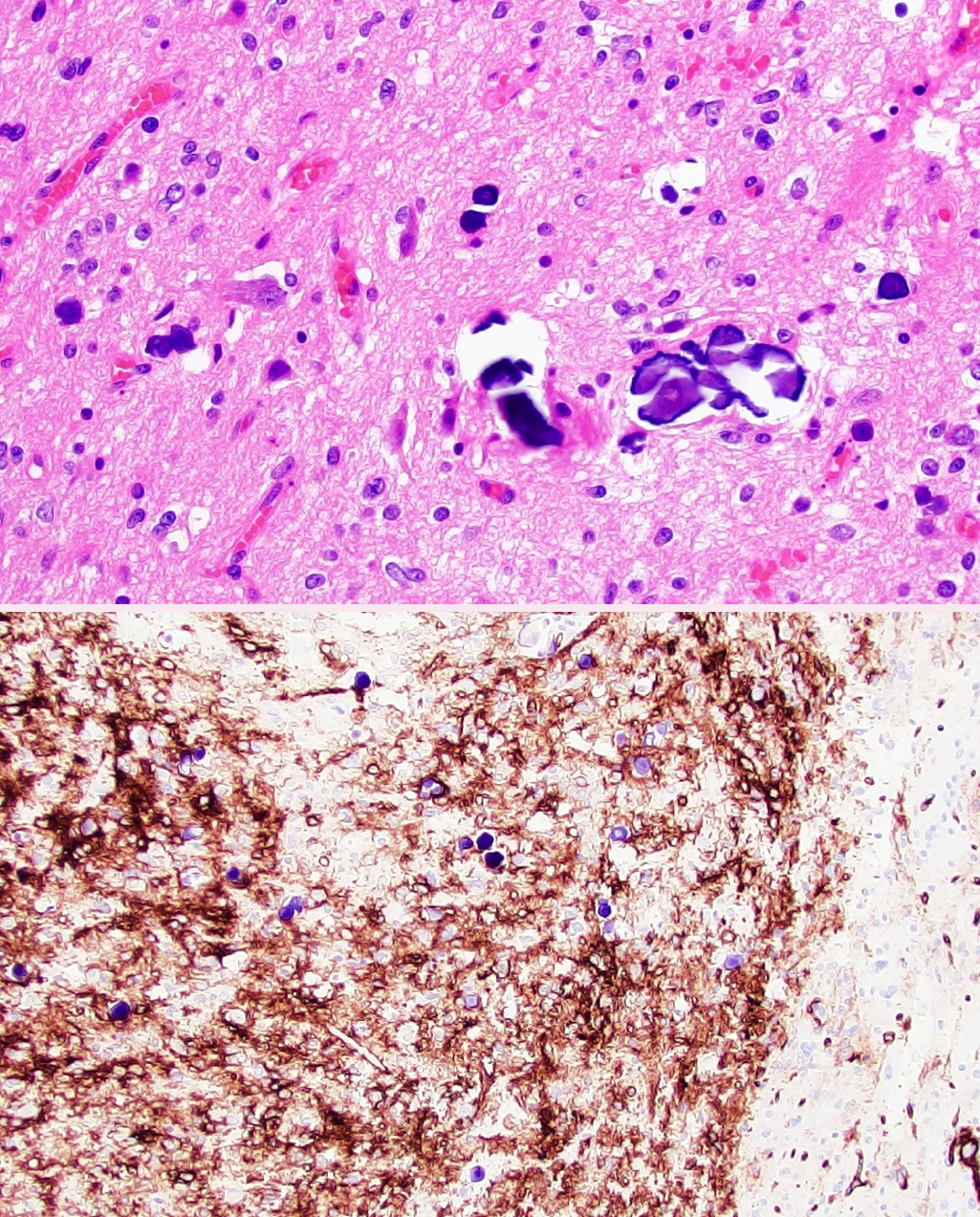
Fig. 2.Myxoid glioneuronal tumor. Small, monomorphic “oligo-like” tumor cells embedded in a prominent myxoid matrix. Molecular characterization of this case revealed a PDGFRA p.K385L mutation.
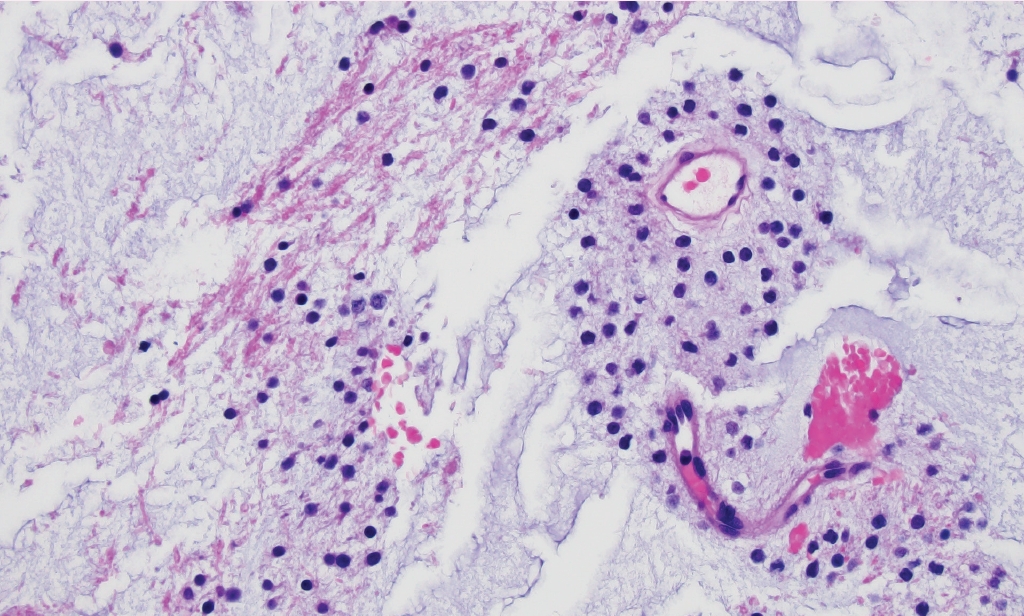
Fig. 3.Primary intracranial sarcoma, DICER1-mutant. Malignant spindle cells arranged in fascicles. Tumor cells in the center with characteristic eosinophilic cytoplasmic globules.

Fig. 4.Desmoplastic myxoid tumor of the pineal region, SMARCB1-mutant. Histology (top panel) reveals cords of epithelioid tumor cells embedded in a variably myxoid (shown) and desmoplastic (not shown) stroma. Immunohistochemistry demonstrates loss of nuclear SMARCB1 (INI1) expression in tumor cells (bottom panel).
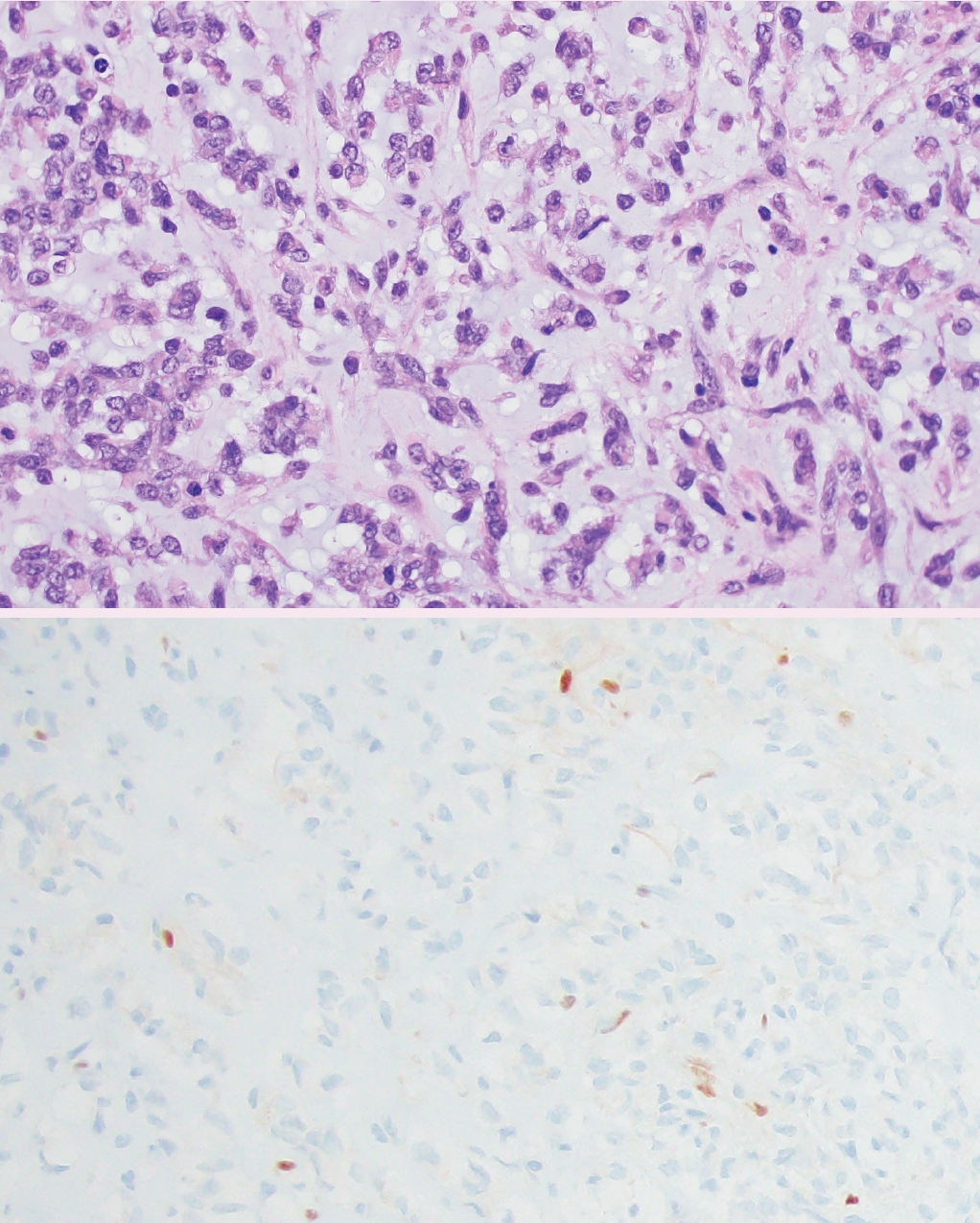
Figure & Data
References
Citations
Citations to this article as recorded by 
- Primary CNS Neuroblastoma, FOXR2‐Activated: Clinicopathological Study of Two Cases With Immunohistochemical Characterization and Literature Review
Sumanta Das, Sunita Ahlawat, Komal Agrawal, Salman Shaikh, Rakesh Kumar Gupta, Suman S. Karanth, Sandeep Vaishya, Rana Patir, Mehar Chand Sharma
Neuropathology.2026;[Epub] CrossRef - Deep Learning for Brain Tumour Analysis: A Systematic Review of CNN‐Transformer Hybrids in Multimodal Imaging
Solomon Buabeng Antwi, Peter Appiahene, Ben Beklisi Kwame Ayawli, Peter Nimbe, Vincenzo Positano
International Journal of Biomedical Imaging.2026;[Epub] CrossRef - Bioinformatics insights into ACSL1 and ACSL5: prognostic and immune roles in low-grade glioma
Cheng Zhang, Zhonghua Lv, Hongsheng Liang, Fulan Hu, Haoran Bi
BMC Cancer.2025;[Epub] CrossRef - Current Understanding of the Exosomes and Their Associated Biomolecules in the Glioblastoma Biology, Clinical Treatment, and Diagnosis
Aghdas Ramezani, Maryam Rahnama, Fatemeh Mahmoudian, Fatemeh Shirazi, Mahmoud Ganji, Shohreh Bakhshi, Bahman Khalesi, Zahra Sadat Hashemi, Saeed Khalili
Journal of Neuroimmune Pharmacology.2025;[Epub] CrossRef - Diagnostic Utility of Intratumoral Susceptibility Signals in Adult Diffuse Gliomas: Tumor Grade Prediction and Correlation with Molecular Markers Within the WHO CNS5 (2021) Classification
José Ignacio Tudela Martínez, Victoria Vázquez Sáez, Guillermo Carbonell, Héctor Rodrigo Lara, Florentina Guzmán-Aroca, Juan de Dios Berna Mestre
Journal of Clinical Medicine.2025; 14(11): 4004. CrossRef - Glioblastoma in Puerto Rico: A 21-year population-based study
Carlos E Calderon-Valero, Esteban Rivera, Odaly Balasquide, Alejandro E Cedeño-Moran, Aixa De Jesus, Miguel Mayol Del Valle
Neuro-Oncology Advances.2025;[Epub] CrossRef - Brain Tumors, AI and Psychiatry: Predicting Tumor-Associated Psychiatric Syndromes with Machine Learning and Biomarkers
Matei Șerban, Corneliu Toader, Răzvan-Adrian Covache-Busuioc
International Journal of Molecular Sciences.2025; 26(17): 8114. CrossRef - Engineered bacteria/bacterial components strategy for glioma
Yan Zhu, Meilin Shen, Qi Chen, Huanghao Yang
Chemical Engineering Journal.2025; 525: 170539. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-




- Related articles
-
- What’s new in hematopathology 2025: myeloid neoplasms in the WHO 5th edition and ICC
- What’s new in adrenal gland pathology: WHO 5th edition for adrenal cortex
- What’s new in thyroid pathology 2024: updates from the new WHO classification and Bethesda system
- What’s new in genitourinary pathology 2023: WHO 5th edition updates for urinary tract, prostate, testis, and penis
- What’s new in dermatopathology 2023: WHO 5th edition updates
What’s new in neuropathology 2024: CNS WHO 5th edition updates
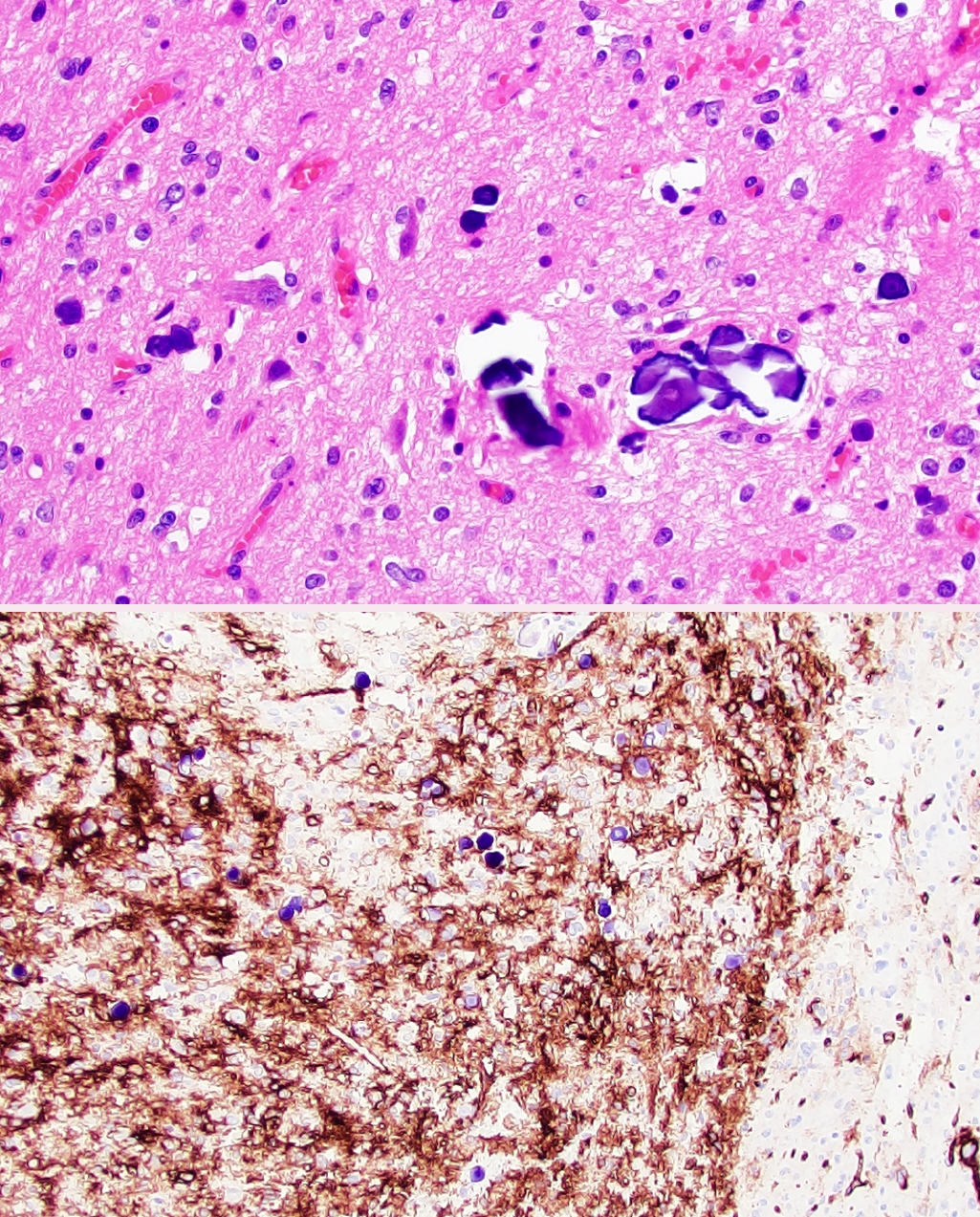

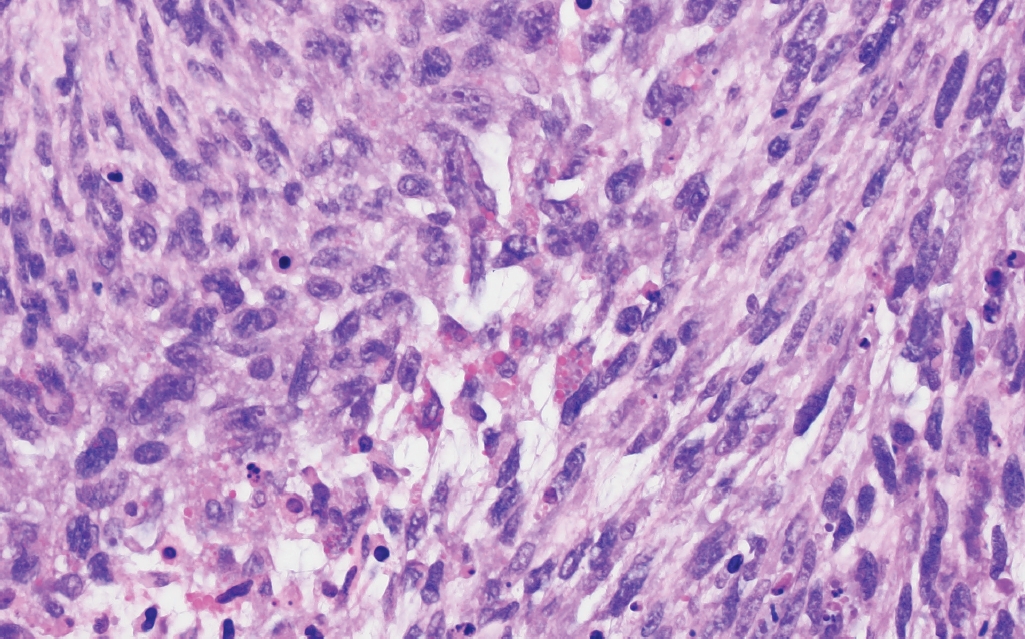
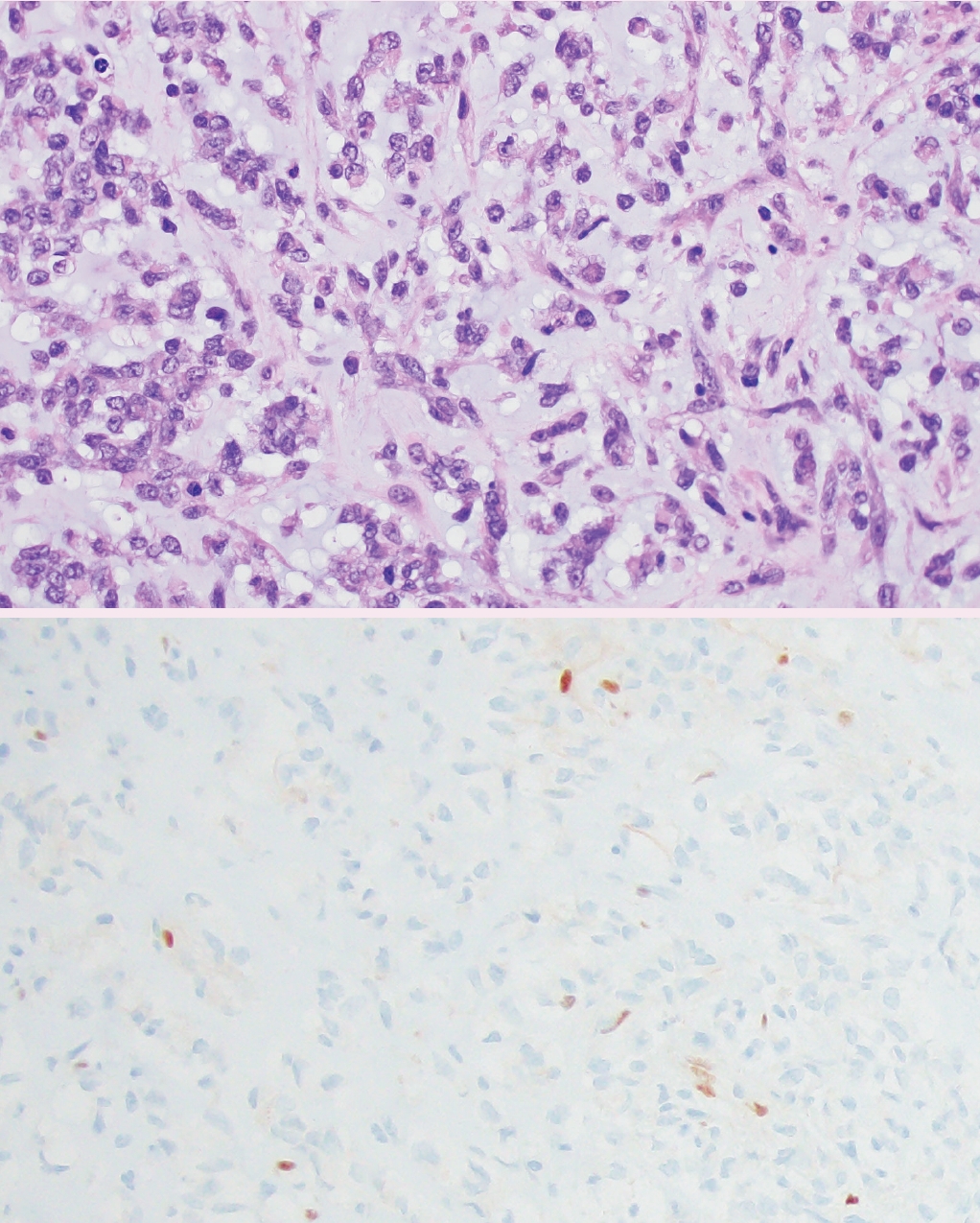
Fig. 1. Polymorphous low-grade neuroepithelial tumor of the young (PLNTY). Low-grade neoplasm demonstrating diffuse growth pattern and microcalcifications (top panel). Strong, regional CD34 expression by tumor cells (bottom panel).
Fig. 2. Myxoid glioneuronal tumor. Small, monomorphic “oligo-like” tumor cells embedded in a prominent myxoid matrix. Molecular characterization of this case revealed a PDGFRA p.K385L mutation.
Fig. 3. Primary intracranial sarcoma, DICER1-mutant. Malignant spindle cells arranged in fascicles. Tumor cells in the center with characteristic eosinophilic cytoplasmic globules.
Fig. 4. Desmoplastic myxoid tumor of the pineal region, SMARCB1-mutant. Histology (top panel) reveals cords of epithelioid tumor cells embedded in a variably myxoid (shown) and desmoplastic (not shown) stroma. Immunohistochemistry demonstrates loss of nuclear SMARCB1 (INI1) expression in tumor cells (bottom panel).
Fig. 1.
Fig. 2.
Fig. 3.
Fig. 4.
What’s new in neuropathology 2024: CNS WHO 5th edition updates

