E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 60(4); 2026 > Article
-
Review Article
Ganglioglioma and gangliocytoma: a review for pathologists -
Gianfranco E. Umeres-Francia
 , Melissa Mejia-Bautista, Pouya Jamshidi, Jared T. Ahrendsen
, Melissa Mejia-Bautista, Pouya Jamshidi, Jared T. Ahrendsen -
Journal of Pathology and Translational Medicine 2026;60(4):379-387.
DOI: https://doi.org/10.4132/jptm.2026.06.06
Published online: July 15, 2026
Department of Pathology, Northwestern University Feinberg School of Medicine, Chicago, IL, USA
- Corresponding Author: Jared T. Ahrendsen, MD, PhD Department of Pathology, Northwestern University Feinberg School of Medicine, 251 East Huron Street, Galter Pavilion 7-132N, Chicago, IL 60611, USA Tel: +1-312-695-0416, Fax: +1-312-503-8249, E-mail: Jared.Ahrendsen@nm.org
This article has been published jointly, with consent, in both Journal of Pathology and Translational Medicine and PathologyOutlines.com.
© The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 44 Views
- 4 Download
Abstract
- Ganglioglioma and gangliocytoma are rare, predominantly low-grade neuroepithelial tumors that commonly present with epilepsy in children and young adults. Advances in molecular profiling have improved understanding of their pathogenesis, highlighting key roles for the mitogen-activated protein kinase/ERK signaling pathway. Diagnosis relies on a combination of clinical, radiologic, and histopathologic features, with complete surgical resection offering the best clinical outcomes. This review summarizes current knowledge on their epidemiology, etiology, clinical presentation, imaging characteristics, pathology, treatment strategies, and prognosis.
- Ganglioglioma and gangliocytoma are rare, low-grade central nervous system (CNS) tumors that primarily affect children and young adults. Patients with these lesions often present with seizures, particularly when located in the temporal lobe. Gangliogliomas are more frequently encountered and are recognized as a leading cause of epilepsy-associated brain tumors in both pediatric and adult populations [1-3]. Gangliocytomas, while less common, share many clinical and histopathological features with gangliogliomas. Despite their relative rarity, these tumors are clinically significant due to their epileptogenic potential and generally favorable outcomes following appropriate management. This review provides a structured overview of gangliogliomas and gangliocytomas, addressing key aspects of their epidemiology, pathogenesis, clinical presentation, neuroimaging characteristics, histopathologic features, molecular properties, therapeutic approaches, and prognostic implications.
INTRODUCTION
- Gangliogliomas have an estimated global incidence of 0.186 cases per 100,000 population [4]. They exhibit a slight male predominance [4-6] and account for approximately 0.4% of all CNS tumors and 1%–7% of pediatric CNS neoplasms [7,8]. These tumors predominantly affect children and young adults, with the majority of cases diagnosed before the age of 20 and a median age of 12 years, although they can occasionally present in older individuals [1]. In epilepsy surgical series, gangliogliomas represent the most frequent histopathological diagnosis among brain tumors associated with intractable epilepsy in adult and pediatric patients, accounting for approximately 40% of such cases [1-3]. Gangliocytomas are much less common, with an incidence of 0.016 cases per 100,000 population [4]. In the context of epilepsy-related surgical resections, gangliocytomas account for approximately 1%–3% of cases [1,9].
EPIDEMIOLOGY
- Gangliogliomas result from genetic alterations that activate the mitogen-activated protein kinase/extracellular-signal-regulated kinase (MAPK/ERK) signaling cascade, including proteins such as MEK, ERK, RSK1, RHEB, and MSK1, which collectively promote aberrant cell proliferation and oncogenesis [10,11]. Although most gangliogliomas occur sporadically, a minority are associated with neurofibromatosis type 1 (NF1) [12]. In addition, gangliogliomas may coexist with adjacent focal cortical dysplasia (FCD), particularly FCD type III [13]. While molecular data regarding gangliocytomas remain limited, their overlapping histopathologic and clinical features with gangliogliomas suggest a shared pathophysiologic basis. Dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) associated with Cowden syndrome is considered a distinct pathologic entity and will not be included in the discussion here (for comprehensive review, see Alanazi et al. [14]). To date, no definitive environmental risk factors have been identified for either tumor type.
ETIOLOGY AND PATHOPHYSIOLOGY
- The clinical presentation of gangliogliomas and gangliocytomas is closely linked to their anatomical location, with lesions in the temporal lobe of the cerebrum frequently associated with seizures and epilepsy. Studies have consistently reported epileptic crises as the most common initial manifestation [5,15], with an average symptom duration of 7.4 years prior to diagnosis [16]. Additional symptoms may include headache, hydrocephalus, and developmental disorders [17].
CLINICAL FEATURES
- Gangliogliomas are typically characterized on brain imaging as circumscribed, cystic masses with an avidly enhancing mural nodule. On magnetic resonance imaging, the cystic component is classically T1 hypointense and T2 hyperintense [18,19]. These tumors often exhibit minimal to no surrounding edema and lack significant mass effect, contributing to their well-delineated appearance. Calcifications are common, present in approximately 40%–50% of cases, and enhancement patterns can vary, especially in less typical presentations where the lesion may appear as a solid mass. The most frequent location is the temporal lobe, followed by the frontal, occipital, and parietal lobes; less commonly, gangliogliomas may arise in the cerebellum, brainstem, or spinal cord [5,18]. Additionally, areas of cortical dysplasia are often observed adjacent to the tumor [18].
RADIOLOGY
- Gross findings
- Gangliogliomas and gangliocytomas are typically grossly well-demarcated lesions, often presenting as variably solid and cystic masses. These tumors characteristically lack areas of hemorrhage and necrosis.
- Frozen section findings
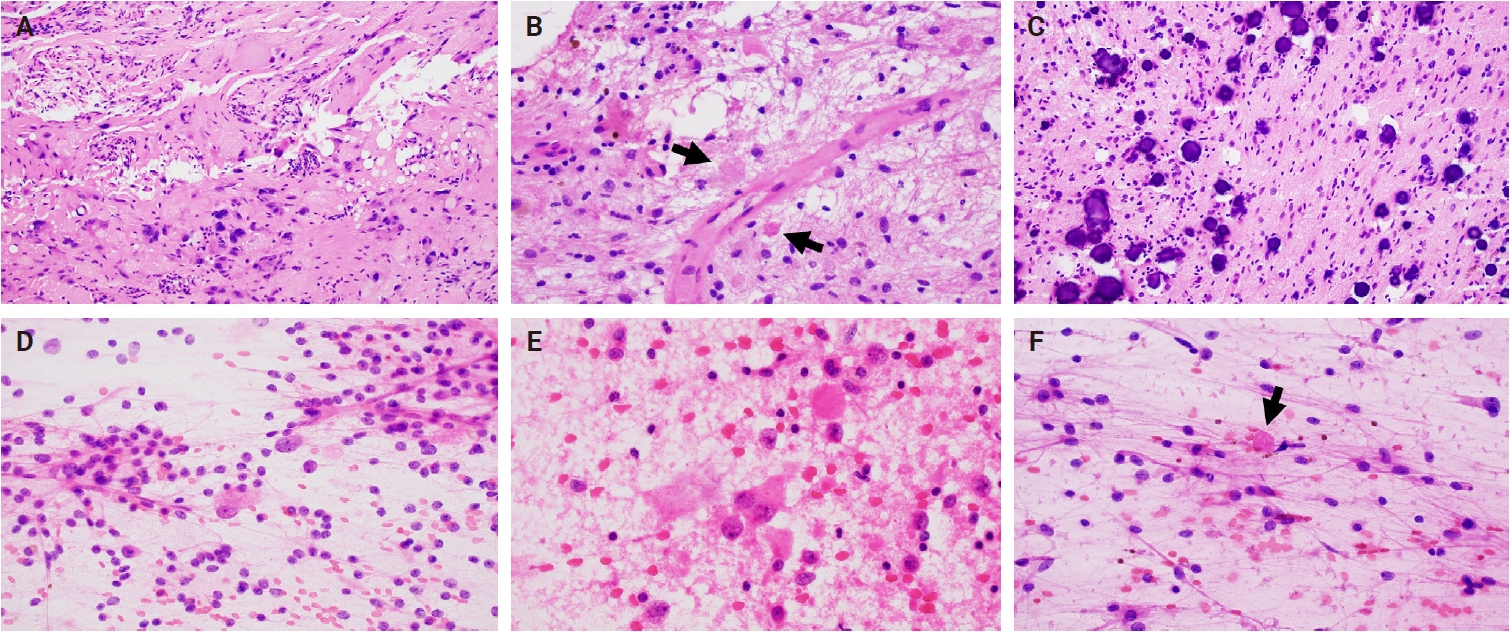
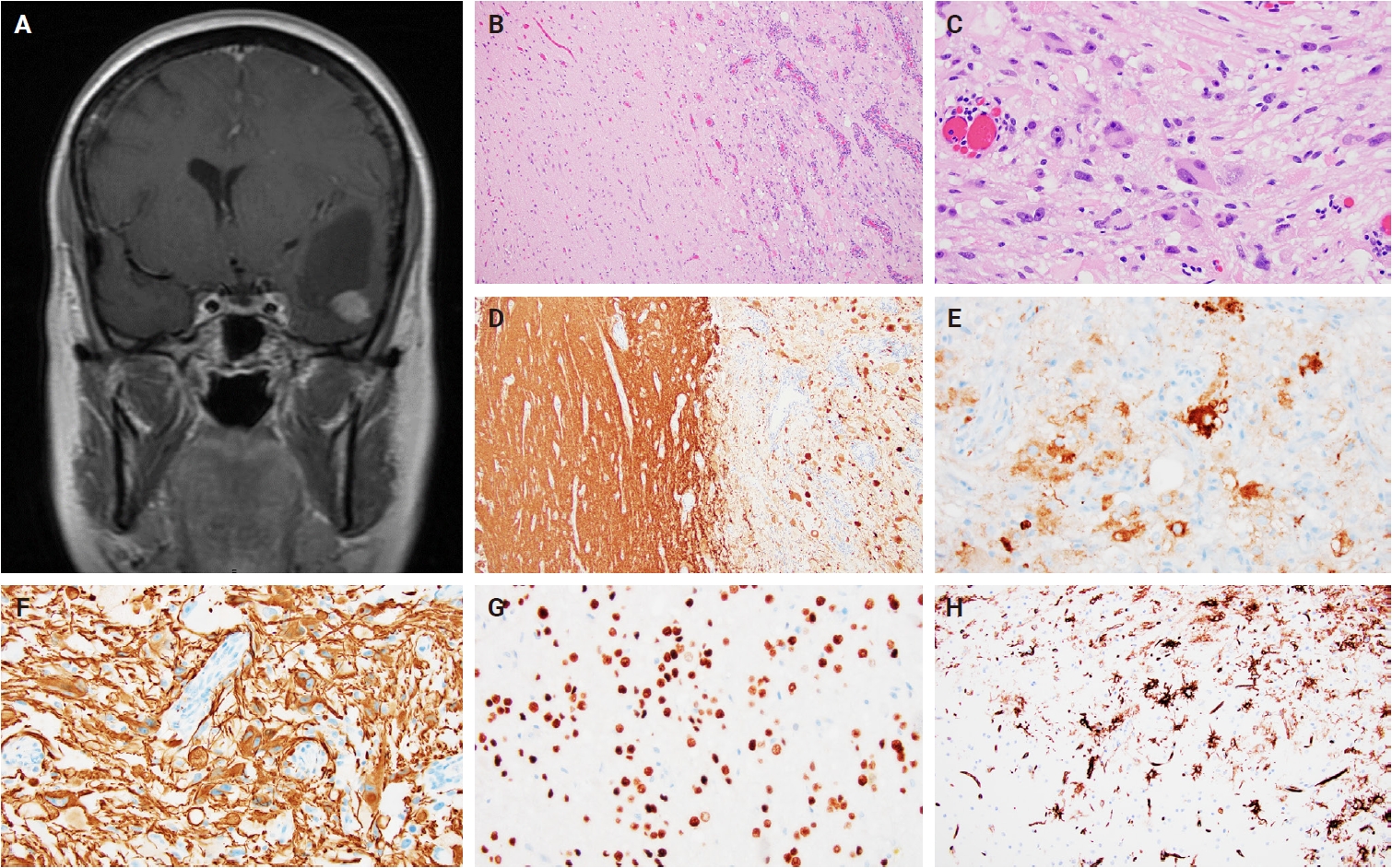
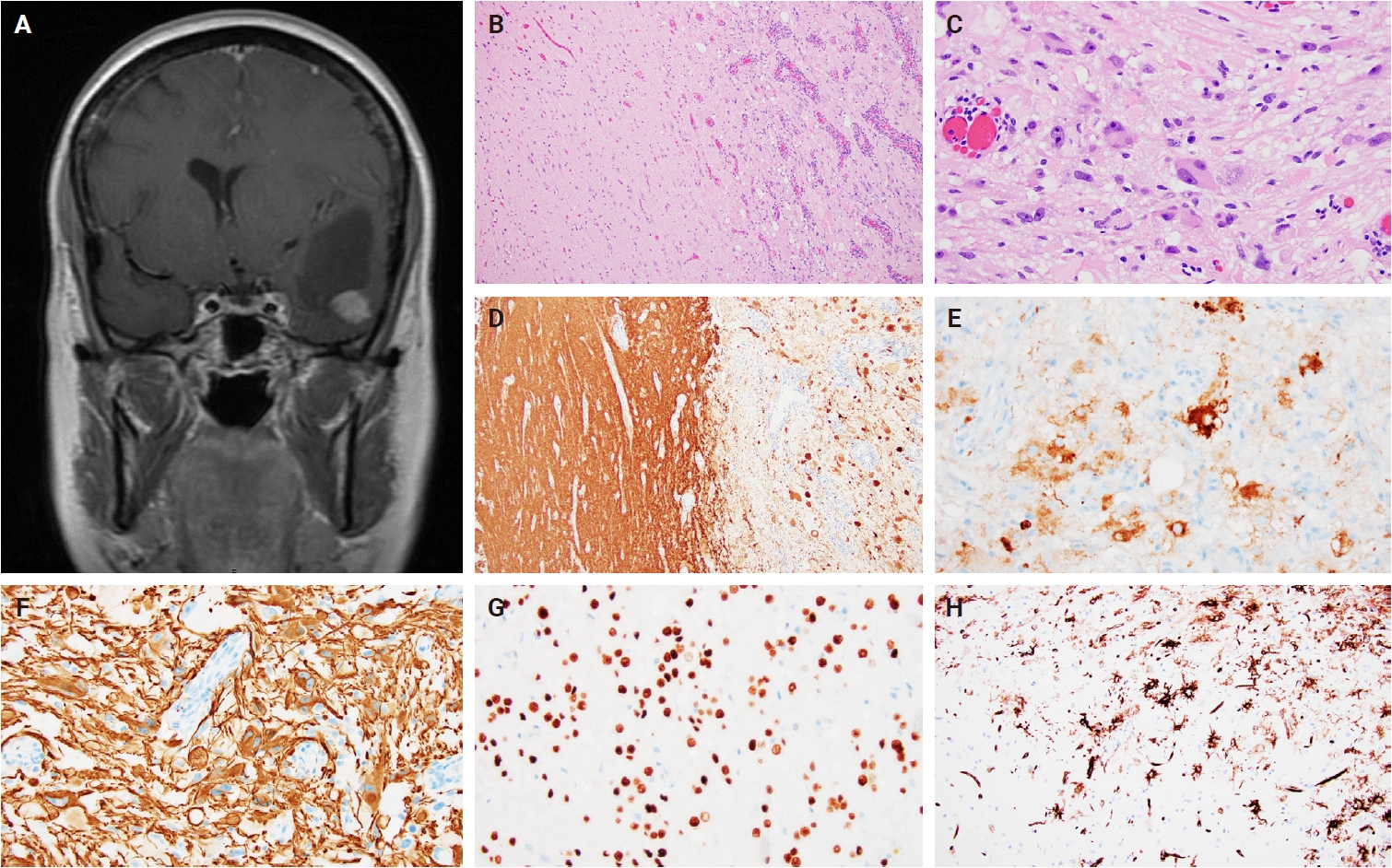
- Intraoperative frozen section evaluation reveals a similarly well-circumscribed mass; however, a diffuse growth pattern involving adjacent brain parenchyma may also be observed, especially at the tumor edge. On a frozen preparation, gangliogliomas exhibit a biphasic cellular composition, comprising dysplastic ganglioid cells intermixed with atypical glial elements (Fig. 1A–C), whereas gangliocytomas are predominantly composed of neuron-like cells. Additional features include variable microcalcifications (Fig. 1C), perivascular lymphocytic infiltrates, and eosinophilic granular bodies (Fig. 1B). Squash preparations of gangliogliomas demonstrate glial cells with fine, piloid-like processes and dysplastic ganglioid cells embedded within a vascular network of thin- to thick-walled vessels (Fig. 1D–F) [20].
- Microscopic findings
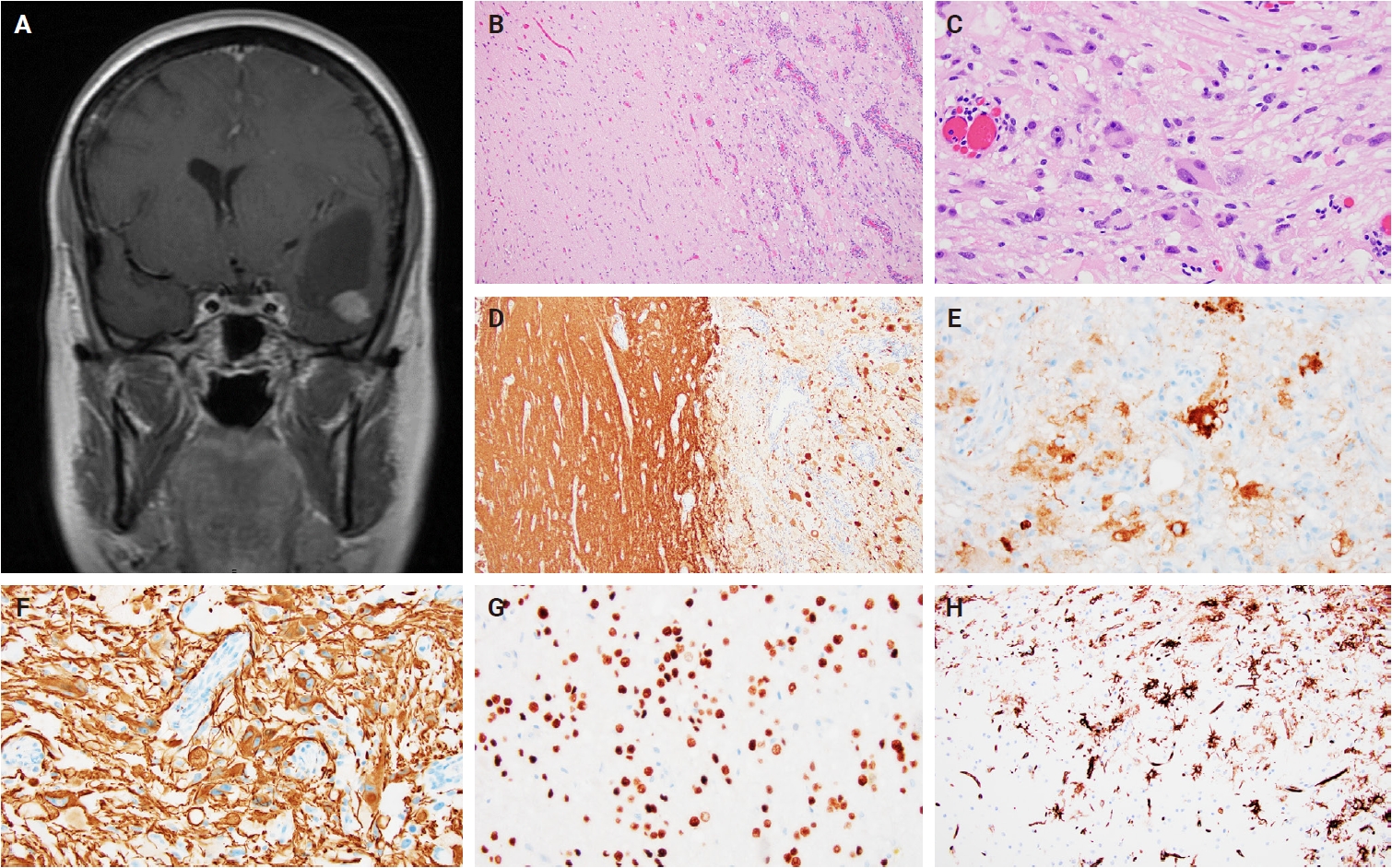
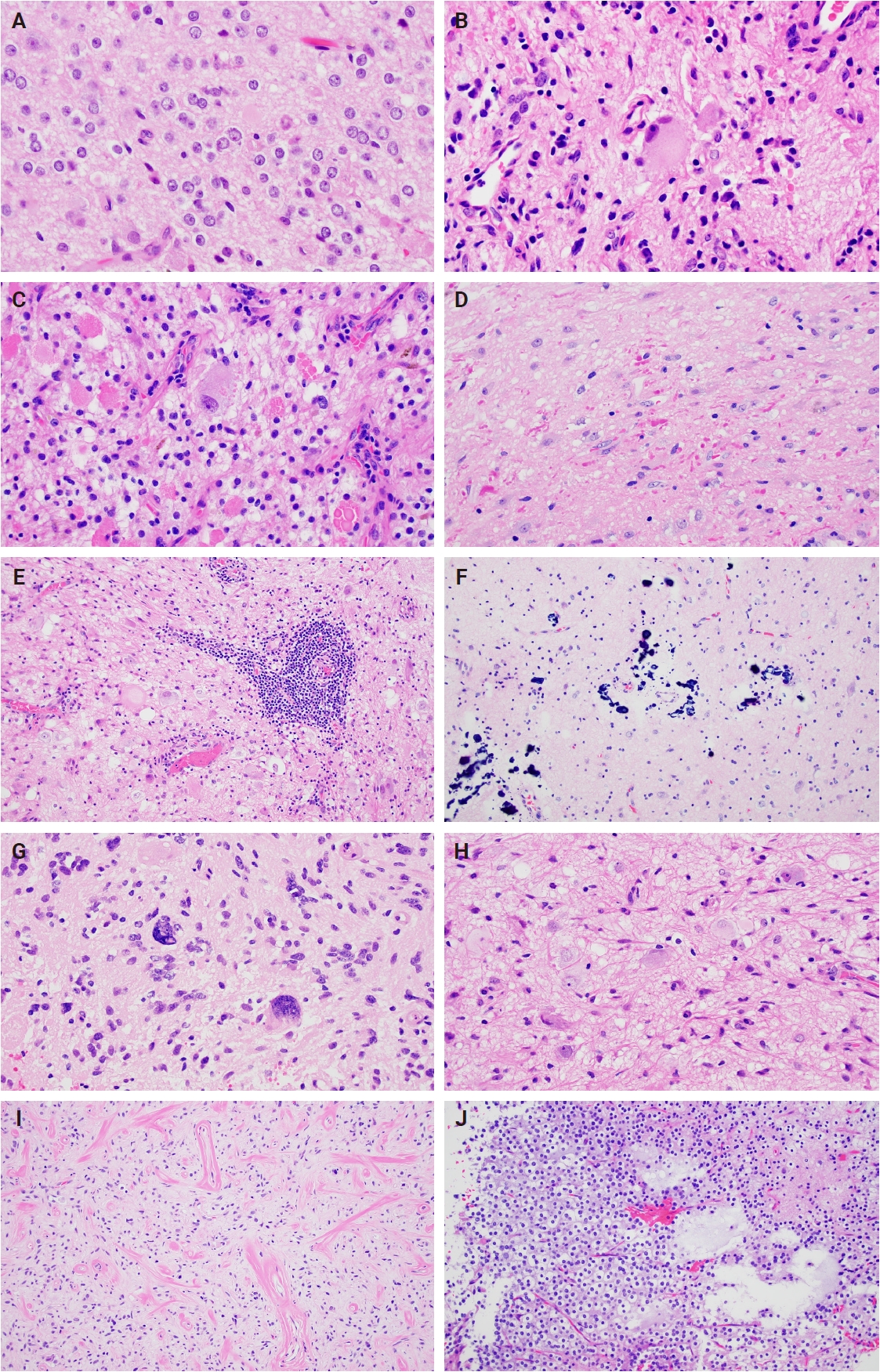
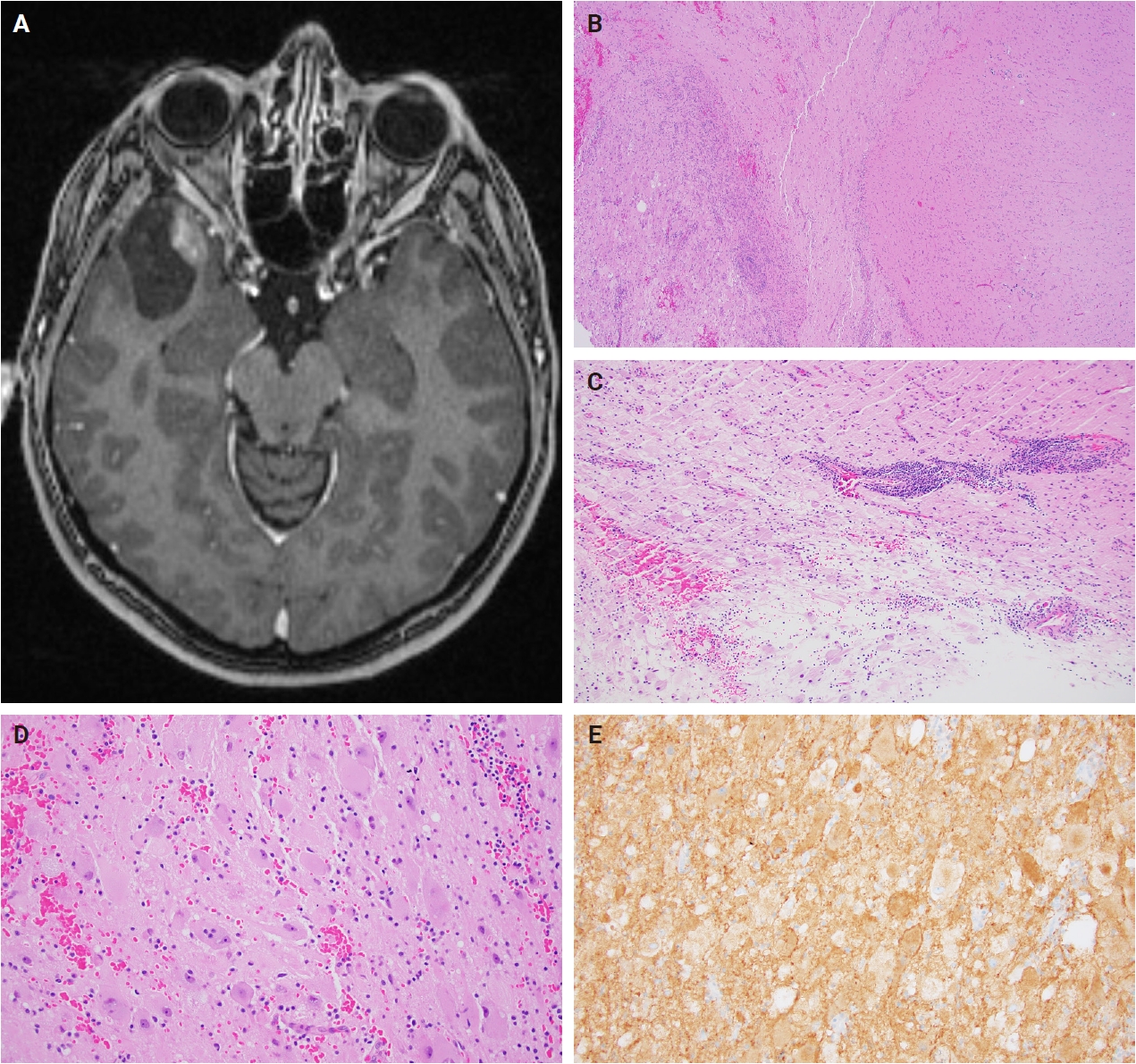
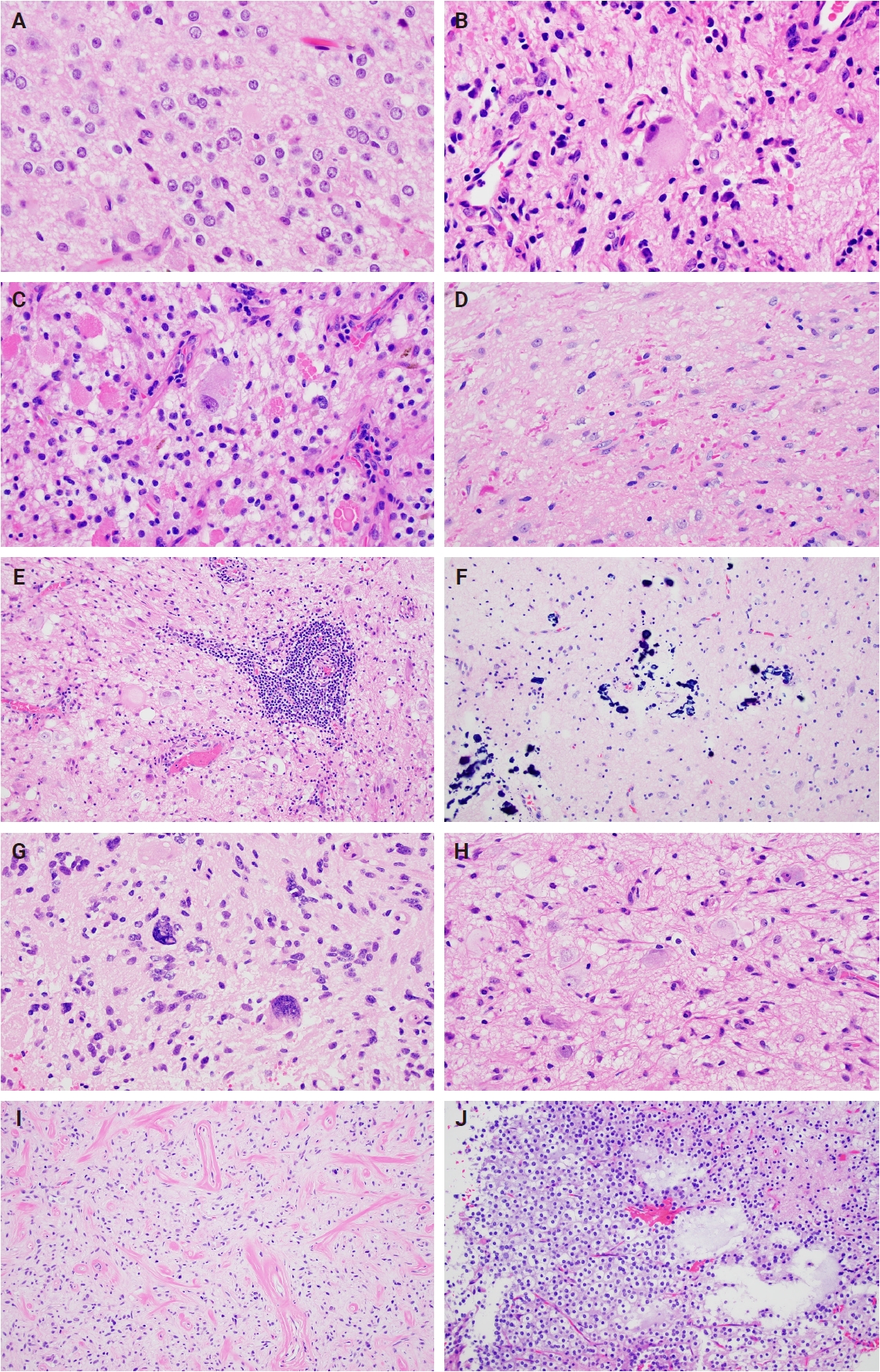
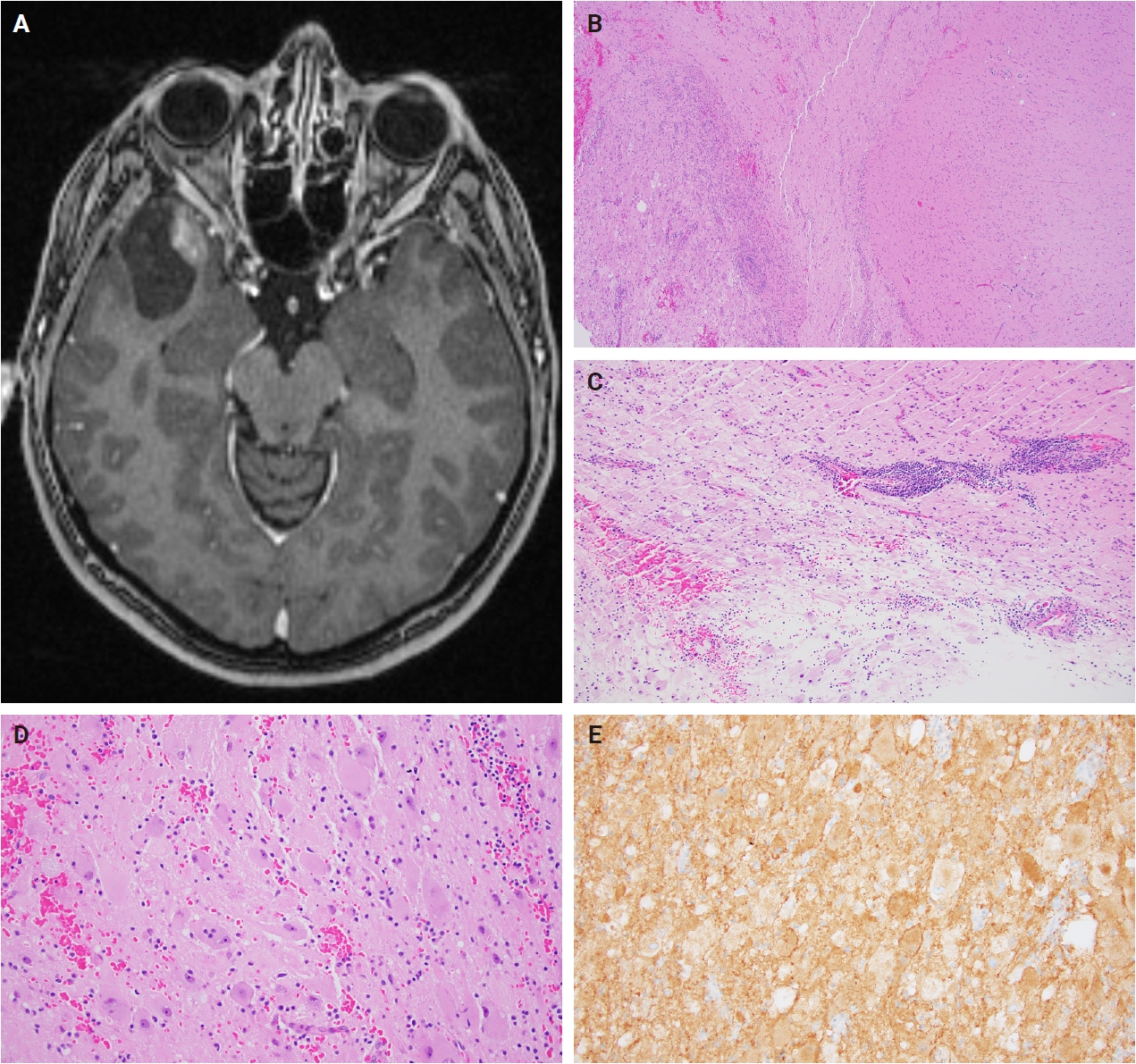
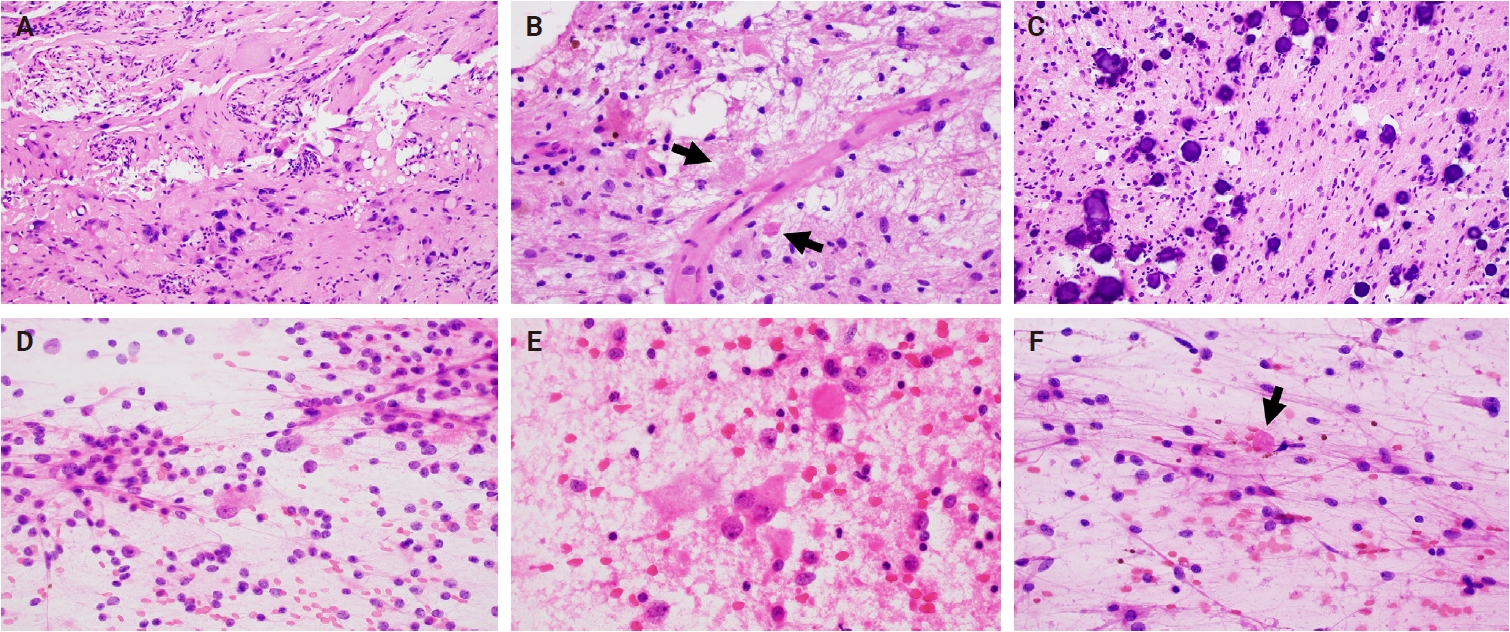
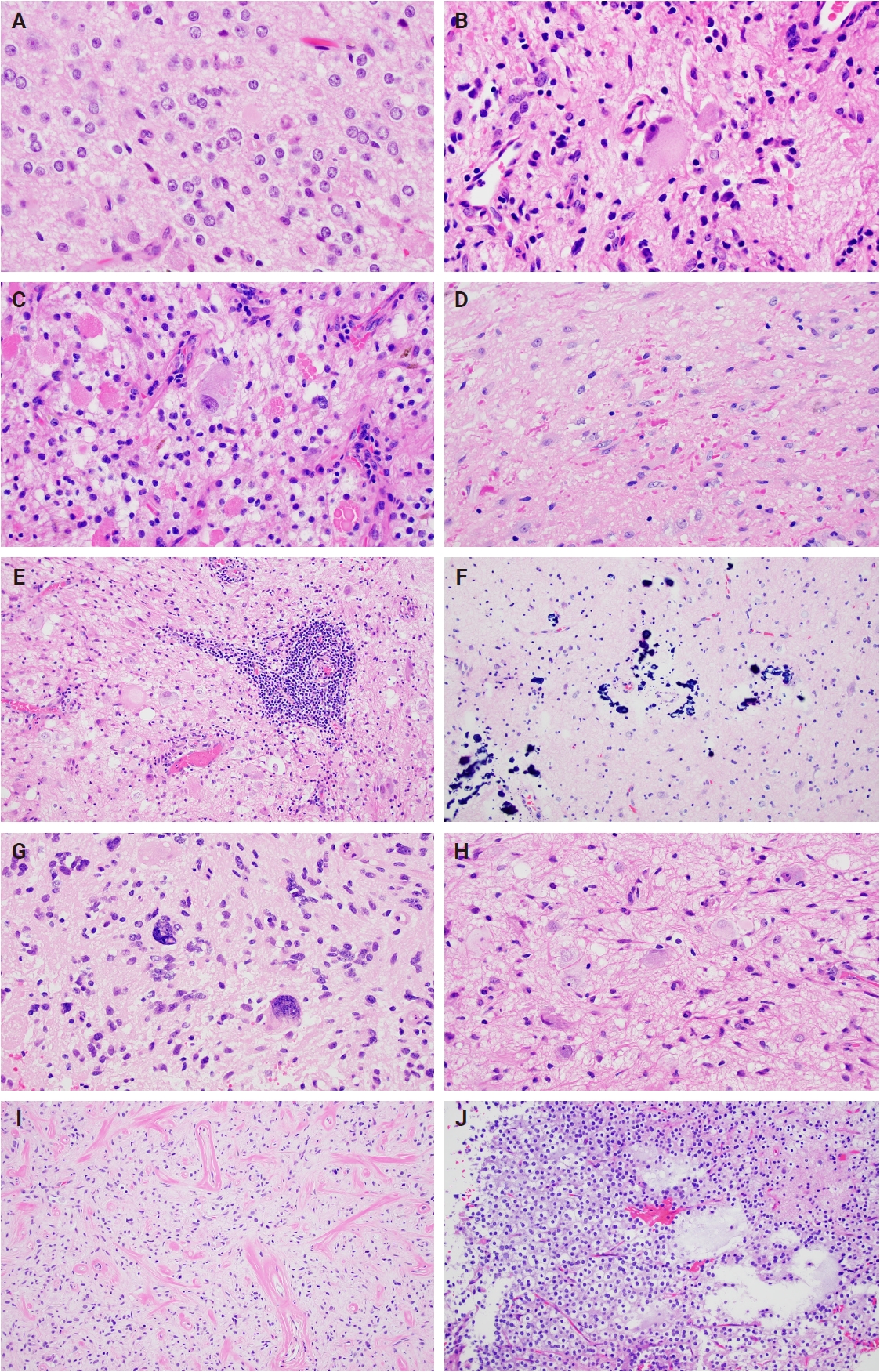
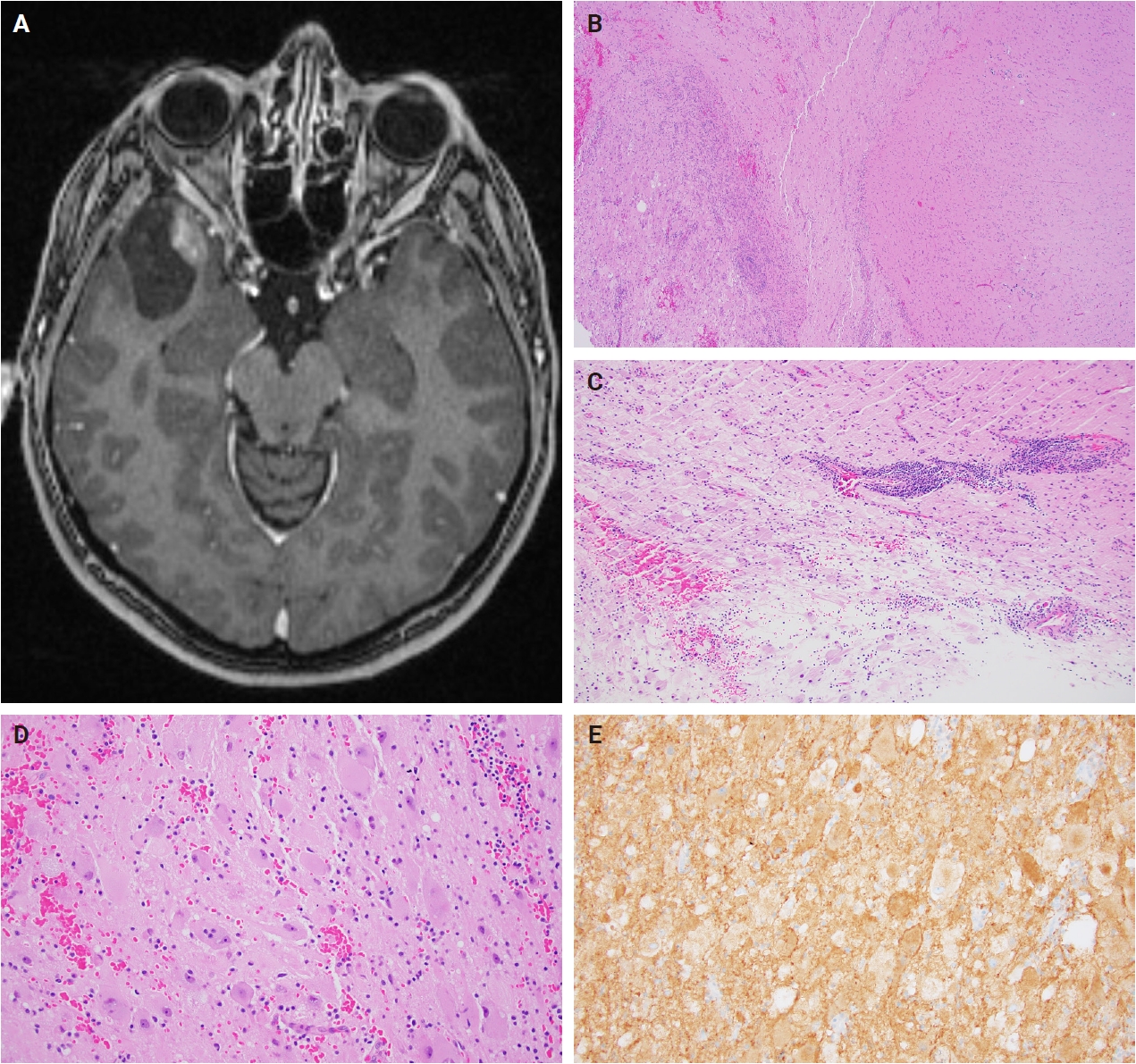
- Histologically, ganglioglioma is a biphasic tumor characterized by a variable admixture of dysmorphic ganglion-like cells and atypical glial elements, generally well-demarcated from the adjacent brain. A representative case with classic histopathologic features is presented in Fig. 2. Microscopically, the dysplastic ganglion-like cells exhibit several hallmark features, including binucleation, cytomegaly with ballooned cytoplasm, and peripheral accumulation of Nissl substance. These cells often form abnormal clusters, referred to as "kissing neurons," and lack an organized cytoarchitecture (Fig. 2C) [5,15]. The glial component comprises hyperchromatic, moderately enlarged cells that may resemble those seen in fibrillary astrocytomas, pilocytic astrocytomas, or oligodendrogliomas [5,21]. Eosinophilic granular bodies are more frequently observed than Rosenthal fibers, and additional features include dystrophic calcifications, perivascular lymphoid infiltrates, and a prominent capillary network. Less common histologic findings include prominent piloid histology, perinuclear halos (i.e., “oligo-like” morphology), and nuclear pleomorphism (Fig. 3). Mitotic activity is typically low to absent [5]. Microscopic infiltration along the tumor edge may be present despite its generally compact growth pattern (Fig. 2B) [1]. Rarely, gangliogliomas may exhibit anaplastic features such as increased mitotic figures, microvascular proliferation, and necrosis. However, the designation “anaplastic ganglioglioma” should be applied cautiously, particularly in the absence of comprehensive molecular characterization to exclude other high-grade gliomas with similar histomorphology, including pleomorphic xanthoastrocytoma [22-24]. In contrast to gangliogliomas, gangliocytomas are composed almost exclusively of large, dysmorphic ganglion cells and lack the neoplastic glial component that characterizes the former; a representative case is presented in Fig. 4.
- Immunohistochemistry
- Ganglion-like cells within gangliogliomas and gangliocytomas express neural markers such as MAP2, neurofilament, and synaptophysin (Figs. 2D, E, 4E). They often lack NeuN immunoreactivity, which is expressed in adjacent non-neoplastic neurons. The glial component of these tumors demonstrates positivity for glial fibrillary acidic protein and Olig2 (Fig. 2F, G) [25]. CD34 expression, either diffuse or patchy in an arborescent pattern, is frequently observed within the tumor or adjacent cortex (Fig. 2H) [26] and may serve as a supportive diagnostic feature in diagnostically challenging cases. Proliferative activity is generally low, with Ki67 indices typically less than 5% [27]. A BRAF V600E mutation–specific antibody is available and often positive in dysplastic ganglion-like cells [28]. These tumors are negative for IDH1 (isocitrate dehydrogenase type 1) R132H and display retained nuclear ATRX expression, distinguishing them from other gliomas with IDH mutations.
PATHOLOGIC FINDINGS
- Gangliogliomas exhibit a diverse range of molecular alterations, with the BRAF V600E mutation being the most common, identified in approximately 10%–60% of cases. Its prevalence is highest in cortical tumors and lowest in spinal cord lesions [21,29,30]. Additional genetic abnormalities include other BRAF mutations, BRAF gene fusions (most frequently KIAA1549::BRAF), KRAS activating mutations, and NF1 inactivating mutations or deletions [31]. The BRAF V600E mutation can be detected via immunohistochemistry or molecular techniques such as gene sequencing, including droplet digital polymerase chain reaction [28]. In cases negative for BRAF V600E, broader molecular testing using next-generation sequencing or gene fusion panels is recommended to identify other MAPK pathway–activating alterations [31]. Furthermore, gangliogliomas demonstrate a distinct genome-wide DNA methylation profile, which can aid in diagnosis, particularly in histologically ambiguous cases [32]. However, despite the high accuracy of DNA methylation profiling, gangliogliomas may remain difficult to classify, comprising 8.7% of tumors lacking confident methylation-based classification [33]. Gangliocytomas currently lack a defined molecular signature, and no characteristic genetic profile has been published to date.
MOLECULAR AND CYTOGENETICS
- Distinguishing the various cortical-based neuroepithelial lesions requires careful integration of clinical, radiologic, histopathologic, immunohistochemical, and molecular features. FCD typically lacks well-circumscribed tumor architecture and retains NeuN immunoreactivity in dysplastic neurons, with genetic alterations often involving the phosphoinositide 3-kinase/AKT/mammalian target of rapamycin pathway. The presence of MAPK activating alterations (such as BRAF V600E) can be helpful to exclude this entity [13,34]. In contrast, infiltrating low-grade gliomas demonstrate entrapped NeuN-positive neurons within a neoplastic glial population and frequently harbor IDH1/IDH2 mutations. In adolescents and young adults, tumors with diffuse architecture may harbor MAPK pathway alterations (i.e., “Diffuse low-grade glioma, MAPK pathway-altered”), but dysplastic ganglion cells are absent. Polymorphous low-grade neuroepithelial tumor of youth (PLNTY) is characterized by FGFR2 (fibroblast growth factor receptor 2)/FGFR3 (fibroblast growth factor receptor 3) fusions or BRAF V600E mutations, diffuse CD34 positivity, and focally infiltrative oligodendroglial-like components [35], though significant overlap may exist between PLNTY and ganglioglioma. Dysembryoplastic neuroepithelial tumors show multinodular architecture with floating neurons in a mucinous matrix and frequent FGFR1 alterations and typically lack CD34 expression [36]. Multinodular vacuolating neural tumors also exhibit multinodular growth, prominent vacuolization, and Olig2-positive neuronal cells, but lack dysmorphic neurons and CD34 staining [37]. Pleomorphic xanthoastrocytoma presents with xanthomatous cells, nuclear pleomorphism, and a reticulin-rich stroma, often harboring both BRAF V600E mutation and CDKN2A/CDKN2B homozygous deletion [38]. Desmoplastic infantile ganglioglioma is distinguished by its large size in infantile patients and a markedly desmoplastic stroma [39]. Hypothalamic hamartomas, pathognomonic for Pallister-Hall syndrome, are congenital lesions presenting with gelastic seizures and GLI3 mutations [40,41]. Lastly, dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) is associated with Cowden syndrome and PTEN loss and lacks MAPK alterations typical of gangliogliomas [42].
DIFFERENTIAL DIAGNOSIS
- The management of gangliogliomas and gangliocytomas centers on surgical intervention, with complete resection representing the cornerstone of treatment and the most significant prognostic factor, particularly for gangliogliomas [7]. Gangliogliomas are low-grade tumors associated with excellent long-term outcomes, with reported 15-year overall survival rates ranging from 83% to 94%. In cases that are progressive, recurrent, or refractory, targeted therapies such as RAF or MEK inhibitors have shown potential benefit, although their clinical utility remains under investigation [43-46]. Emerging molecular data suggest that certain genetic profiles, including co-occurrence of BRAF V600E with CDKN2A/CDKN2B homozygous deletions or midline tumors harboring both BRAF V600E and H3 K27M mutations, may be associated with poorer outcomes [31,47-49]. Another report suggests tumors with PTPN11/KRAS/NF1 and other MAPK pathway alterations may show atypical histologic features and adverse clinical outcomes [50]. Similarly, gangliocytomas are benign lesions with excellent long-term survival, and to date, specific prognostic markers have not been identified.
TREATMENT AND PROGNOSIS
- Gangliogliomas and gangliocytomas represent a distinct subset of neuroepithelial tumors with unique clinical, radiologic, and pathologic features. While generally associated with favorable outcomes, particularly when amenable to complete surgical resection, their biological behavior can vary depending on underlying molecular alterations and anatomical location. Advances in molecular profiling have enhanced our understanding of their pathogenesis, revealing key roles for MAPK/ERK signaling pathway abnormalities. These findings have prompted exploration of targeted therapies in select cases, though their effectiveness and long-term outcomes remain uncertain. Through this review, we have highlighted the current knowledge surrounding these tumors, aiming to support clinicians and researchers in the comprehensive evaluation and management of gangliogliomas and gangliocytomas.
CONCLUSION
Ethics Statement
Not applicable.
Availability of Data and Material
Data sharing not applicable to this article as no datasets were generated or analyzed during the study.
Code Availability
Not applicable.
Author Contributions
Conceptualization: JTA. Supervision: JTA. Writing—original draft: all authors. Approval of final manuscript: all authors.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
Funding Statement
No funding to declare.
- 1. Blumcke I, Spreafico R, Haaker G, et al. Histopathological findings in brain tissue obtained during epilepsy surgery. N Engl J Med 2017; 377: 1648-56. ArticlePubMed
- 2. Prayson RA. Tumours arising in the setting of paediatric chronic epilepsy. Pathology 2010; 42: 426-31. ArticlePubMed
- 3. Prayson RA. Brain tumors in adults with medically intractable epilepsy. Am J Clin Pathol 2011; 136: 557-63. ArticlePubMed
- 4. Darlix A, Zouaoui S, Rigau V, et al. Epidemiology for primary brain tumors: a nationwide population-based study. J Neurooncol 2017; 131: 525-46. ArticlePubMedPDF
- 5. Blumcke I, Wiestler OD. Gangliogliomas: an intriguing tumor entity associated with focal epilepsies. J Neuropathol Exp Neurol 2002; 61: 575-84. ArticlePubMed
- 6. Yang I, Chang EF, Han SJ, et al. Early surgical intervention in adult patients with ganglioglioma is associated with improved clinical seizure outcomes. J Clin Neurosci 2011; 18: 29-33. ArticlePubMed
- 7. Compton JJ, Laack NN, Eckel LJ, Schomas DA, Giannini C, Meyer FB. Long-term outcomes for low-grade intracranial ganglioglioma: 30-year experience from the Mayo Clinic. J Neurosurg 2012; 117: 825-30. ArticlePubMed
- 8. Luyken C, Blumcke I, Fimmers R, Urbach H, Wiestler OD, Schramm J. Supratentorial gangliogliomas: histopathologic grading and tumor recurrence in 184 patients with a median follow-up of 8 years. Cancer 2004; 101: 146-55. ArticlePubMed
- 9. Thom M, Blumcke I, Aronica E. Long-term epilepsy-associated tumors. Brain Pathol 2012; 22: 350-79. ArticlePubMedPMC
- 10. Aronica E, Boer K, Doorn KJ, et al. Expression and localization of voltage dependent potassium channel Kv4.2 in epilepsy associated focal lesions. Neurobiol Dis 2009; 36: 81-95. ArticlePubMed
- 11. Rak B, Szlufik S, Grajkowska W, et al. Upregulation of mitogen-activated protein kinase in ganglioglioma. Folia Neuropathol 2013; 51: 283-9. ArticlePubMed
- 12. Rodriguez FJ, Perry A, Gutmann DH, et al. Gliomas in neurofibromatosis type 1: a clinicopathologic study of 100 patients. J Neuropathol Exp Neurol 2008; 67: 240-9. ArticlePubMedPMC
- 13. Blumcke I, Thom M, Aronica E, et al. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia 2011; 52: 158-74. ArticlePubMedPMC
- 14. Alanazi AI, Alanezi T, Aljofan ZF, Alarabi A, Elwatidy S. Lhermitte-Duclos disease: a systematic review. Surg Neurol Int 2023; 14: 351.ArticlePubMedPMC
- 15. Wolf HK, Muller MB, Spanle M, Zentner J, Schramm J, Wiestler OD. Ganglioglioma: a detailed histopathological and immunohistochemical analysis of 61 cases. Acta Neuropathol 1994; 88: 166-73. ArticlePubMedPDF
- 16. Gelabert-Gonzalez M, Amo JM, Arcos Algaba A, et al. Intracranial gangliogliomas: a review of a series of 20 patients. Neurologia 2011; 26: 405-15. ArticlePubMed
- 17. Leclerc A, Le Hello-Regnier E, Faisant M, et al. Ganglioglioma revealed by spontaneous intracerebral hematoma: a cohort study. Neurochirurgie 2022; 68: e8-15. ArticlePubMed
- 18. Abdel Razek AA, Elsebaie NA, Zamora C, Castillo M. Imaging of neuronal and mixed glioneuronal tumors. J Comput Assist Tomogr 2020; 44: 356-69. ArticlePubMed
- 19. Shin JH, Lee HK, Khang SK, et al. Neuronal tumors of the central nervous system: radiologic findings and pathologic correlation. Radiographics 2002; 22: 1177-89. ArticlePubMed
- 20. Kurtulan O, Bilginer B, Soylemezoglu F. Challenges in the intraoperative consultation of low-grade epilepsy-associated neuroepithelial tumors by cytomorphology in squash preparations. Acta Cytol 2022; 66: 142-8. ArticlePubMedPDF
- 21. Gessi M, Dorner E, Dreschmann V, et al. Intramedullary gangliogliomas: histopathologic and molecular features of 25 cases. Hum Pathol 2016; 49: 107-13. ArticlePubMed
- 22. Reinhardt A, Pfister K, Schrimpf D, et al. Anaplastic ganglioglioma: a diagnosis comprising several distinct tumour types. Neuropathol Appl Neurobiol 2022; 48: e12847. ArticlePubMed
- 23. Terrier LM, Bauchet L, Rigau V, et al. Natural course and prognosis of anaplastic gangliogliomas: a multicenter retrospective study of 43 cases from the French Brain Tumor Database. Neuro Oncol 2017; 19: 678-88. ArticlePubMedPMC
- 24. Zanello M, Pages M, Tauziede-Espariat A, et al. Clinical, imaging, histopathological and molecular characterization of anaplastic ganglioglioma. J Neuropathol Exp Neurol 2016; 75: 971-80. ArticlePubMed
- 25. Hirose T, Scheithauer BW, Lopes MB, Gerber HA, Altermatt HJ, VandenBerg SR. Ganglioglioma: an ultrastructural and immunohistochemical study. Cancer 1997; 79: 989-1003. ArticlePubMed
- 26. Blumcke I, Giencke K, Wardelmann E, et al. The CD34 epitope is expressed in neoplastic and malformative lesions associated with chronic, focal epilepsies. Acta Neuropathol 1999; 97: 481-90. ArticlePubMedPDF
- 27. Prayson RA, Khajavi K, Comair YG. Cortical architectural abnormalities and MIB1 immunoreactivity in gangliogliomas: a study of 60 patients with intracranial tumors. J Neuropathol Exp Neurol 1995; 54: 513-20. ArticlePubMed
- 28. Koelsche C, Wohrer A, Jeibmann A, et al. Mutant BRAF V600E protein in ganglioglioma is predominantly expressed by neuronal tumor cells. Acta Neuropathol 2013; 125: 891-900. ArticlePubMedPDF
- 29. Qaddoumi I, Orisme W, Wen J, et al. Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol 2016; 131: 833-45. ArticlePubMedPMCPDF
- 30. Schindler G, Capper D, Meyer J, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol 2011; 121: 397-405. ArticlePubMedPDF
- 31. Pekmezci M, Villanueva-Meyer JE, Goode B, et al. The genetic landscape of ganglioglioma. Acta Neuropathol Commun 2018; 6: 47.ArticlePubMedPMCPDF
- 32. Capper D, Jones DT, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature 2018; 555: 469-74. ArticlePubMedPMC
- 33. Drexler R, Brembach F, Sauvigny J, et al. Unclassifiable CNS tumors in DNA methylation-based classification: clinical challenges and prognostic impact. Acta Neuropathol Commun 2024; 12: 9.ArticlePubMedPMCPDF
- 34. Marsan E, Baulac S. Review: mechanistic target of rapamycin (mTOR) pathway, focal cortical dysplasia and epilepsy. Neuropathol Appl Neurobiol 2018; 44: 6-17. ArticlePubMedPDF
- 35. Huse JT, Snuderl M, Jones DT, et al. Polymorphous low-grade neuroepithelial tumor of the young (PLNTY): an epileptogenic neoplasm with oligodendroglioma-like components, aberrant CD34 expression, and genetic alterations involving the MAP kinase pathway. Acta Neuropathol 2017; 133: 417-29. ArticlePubMedPMCPDF
- 36. Blumcke I, Coras R, Wefers AK, et al. Review: challenges in the histopathological classification of ganglioglioma and DNT: microscopic agreement studies and a preliminary genotype-phenotype analysis. Neuropathol Appl Neurobiol 2019; 45: 95-107. ArticlePubMedPDF
- 37. Pekmezci M, Stevers M, Phillips JJ, et al. Multinodular and vacuolating neuronal tumor of the cerebrum is a clonal neoplasm defined by genetic alterations that activate the MAP kinase signaling pathway. Acta Neuropathol 2018; 135: 485-8. ArticlePubMedPMCPDF
- 38. Phillips JJ, Gong H, Chen K, et al. The genetic landscape of anaplastic pleomorphic xanthoastrocytoma. Brain Pathol 2019; 29: 85-96. ArticlePubMedPDF
- 39. VandenBerg SR. Desmoplastic infantile ganglioglioma and desmoplastic cerebral astrocytoma of infancy. Brain Pathol 1993; 3: 275-81. ArticlePubMed
- 40. Demurger F, Ichkou A, Mougou-Zerelli S, et al. New insights into genotype-phenotype correlation for GLI3 mutations. Eur J Hum Genet 2015; 23: 92-102. ArticlePubMed
- 41. Dunham C, McFadden D, Dahlgren L, Butler B, Hamilton S, McKinnon M. Congenital hypothalamic "hamartoblastoma" versus "hamartoma": suggestions for neuropathologic terminology emanating from a mid-gestational autopsy case of Pallister-Hall syndrome. Pediatr Dev Pathol 2018; 21: 324-31. ArticlePubMedPDF
- 42. Riegert-Johnson DL, Gleeson FC, Roberts M, et al. Cancer and Lhermitte-Duclos disease are common in Cowden syndrome patients. Hered Cancer Clin Pract 2010; 8: 6.ArticlePubMedPMCPDF
- 43. Berzero G, Bellu L, Baldini C, et al. Sustained tumor control with MAPK inhibition in BRAF V600-mutant adult glial and glioneuronal tumors. Neurology 2021; 97: e673-83. ArticlePubMed
- 44. del Bufalo F, Carai A, Figa-Talamanca L, et al. Response of recurrent BRAFV600E mutated ganglioglioma to vemurafenib as single agent. J Transl Med 2014; 12: 356.ArticlePubMedPMC
- 45. Fusco MJ, Pina Y, Macaulay RJ, et al. Durable progression-free survival with the use of BRAF and MEK inhibitors in four cases with BRAF V600E-mutated gliomas. Cancer Control 2021; 28: 10732748211040013.ArticlePubMedPMCPDF
- 46. Rush S, Foreman N, Liu A. Brainstem ganglioglioma successfully treated with vemurafenib. J Clin Oncol 2013; 31: e159-60. ArticlePubMed
- 47. Lassaletta A, Zapotocky M, Mistry M, et al. Therapeutic and prognostic implications of BRAF V600E in pediatric low-grade gliomas. J Clin Oncol 2017; 35: 2934-41. ArticlePubMedPMC
- 48. Pages M, Beccaria K, Boddaert N, et al. Co-occurrence of histone H3 K27M and BRAF V600E mutations in paediatric midline grade I ganglioglioma. Brain Pathol 2018; 28: 103-11. ArticlePubMedPMCPDF
- 49. Ryall S, Zapotocky M, Fukuoka K, et al. Integrated molecular and clinical analysis of 1,000 pediatric low-grade gliomas. Cancer Cell 2020; 37: 569-83. ArticlePubMedPMC
- 50. Hoffmann L, Coras R, Kobow K, et al. Ganglioglioma with adverse clinical outcome and atypical histopathological features were defined by alterations in PTPN11/KRAS/NF1 and other RAS-/MAP-Kinase pathway genes. Acta Neuropathol 2023; 145: 815-27. ArticlePubMedPMCPDF
REFERENCES
Figure & Data
References
Citations
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-