 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 60(4); 2026 > Article
-
Review Article
Angiomatoid fibrous histiocytoma: a review -
Alexander N. Perez1
 , Phyu P. Aung2
, Phyu P. Aung2 -
Journal of Pathology and Translational Medicine 2026;60(4):371-378.
DOI: https://doi.org/10.4132/jptm.2026.06.05
Published online: July 15, 2026
1Department of Pathology, Microbiology, and Immunology, Vanderbilt University Medical Center, Nashville, TN, USA
2Department of Pathology and Laboratory Medicine, University of Texas MD Anderson Cancer Center, Houston, TX, USA
- Corresponding Author: Alexander N. Perez, MD Department of Pathology, Microbiology, and Immunology, Vanderbilt University Medical Center, 445 Great Circle Rd, Office 1959, Nashville, TN 37228, USA Tel: +1-615-875-1500, Fax: +1-615-343-7023, E-mail: alexander.n.perez@vumc.org
This article has been published jointly, with consent, in both Journal of Pathology and Translational Medicine and PathologyOutlines.com.
© The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 35 Views
- 1 Download
Abstract
- Angiomatoid fibrous histiocytoma is a rare mesenchymal neoplasm of uncertain cell lineage with indeterminate behavior, hallmarked by EWSR1 translocations. This tumor typically arises in the subcutaneous or deep soft tissues and is composed of bland to mildly atypical histiocytoid cells with frequent intralesional hemorrhagic pseudocystic spaces. It affects both children and adults, without a significant sex predilection. Histologically, the tumor may be mistaken for a lymph node given the apparent predilection for node-bearing sites as well as the brisk lymphoid cuff featuring germinal centers. Surgical excision is often curative, with local recurrence occurring occasionally and metastasis only very rarely. A possible relationship to molecularly related entities arising primarily within the thoracic cavity and intracranial compartment has been proposed, although this association remains incompletely understood.
- Angiomatoid fibrous histiocytoma (AFH), first described as “Angiomatoid malignant fibrous histiocytoma,” is an ultra-rare tumor of uncertain lineage [1] and is categorized by the World Health Organization as an intermediate tumor of uncertain differentiation [2]. At the time of its initial description by Enzinger in 1979 [1], it was considered a variant of what was then known as “malignant fibrous histiocytoma” (now undifferentiated pleomorphic sarcoma). However, the predominantly younger age distribution, significantly favorable prognosis, and subsequently identified recurrent molecular alteration(s) led to the recognition of AFH as a novel/distinct clinicopathologic entity.
- Although AFH is now well established as a distinct tumor type, its exact histogenesis remains uncertain. The lesional cells show some morphologic resemblance to histiocytes but they lack other supporting features (including immunophenotype) indicative of true histiocytic differentiation. Other proposed lines of differentiation have included fibroblastic reticular cell or myoid differentiation; however, no definitive conclusions have been established [3,4]. To date, there is no known precursor lesion or associated predisposing condition linked to the development of AFH.
ETIOLOGY AND PATHOGENESIS
- The clinical and radiologic features of AFH are largely nonspecific, with most patients presenting with a slow-growing, palpable, and typically painless soft tissue mass. Clinically, these lesions may raise concern for lymphadenopathy, particularly when arising in node-bearing regions. Radiologic identification of blood-filled spaces with or without fluid-fluid levels may suggest AFH; however, these findings should be interpreted cautiously, as mimickers such as hematoma and vascular proliferations are significantly more common [5].
- AFH occurs most frequently in children and young adults, particularly in patients younger than 30 years of age; however, cases have been reported across a wide age range, including rare congenital presentations [1,6,7]. No significant sex predilection has been identified in several large series [1,4,6]. AFH most commonly presents as a dermal, subcutaneous, or deep soft tissue mass, with the upper and lower extremities representing frequent sites of involvement. A recent study also described a cohort arising specifically in acral locations. Rarely, AFH has been reported as a primary intraosseous neoplasm [8]. Importantly, the anatomic distribution of AFH may overlap with common lymph node–bearing regions, potentially leading to clinical concern for metastatic disease from an occult primary site [9].
- An uncommon but well-documented manifestation of AFH is the development of paraneoplastic inflammatory symptoms related to excess interleukin-6 (IL-6) production by the tumor. In tumors harboring CREB1 (cAMP response element-binding protein 1)–related fusion transcripts, constitutive activation of transcriptional pathways is thought to promote IL-6 overexpression through multiple transcription factor binding sites within the IL-6 gene promoter region [10-13]. Associated systemic manifestations may include fever, weight loss, anemia, malaise, and other “B symptoms,” resembling those encountered in other neoplastic and inflammatory disorders with paraneoplastic syndromes [10].
CLINICAL FEATURES AND RADIOLOGY
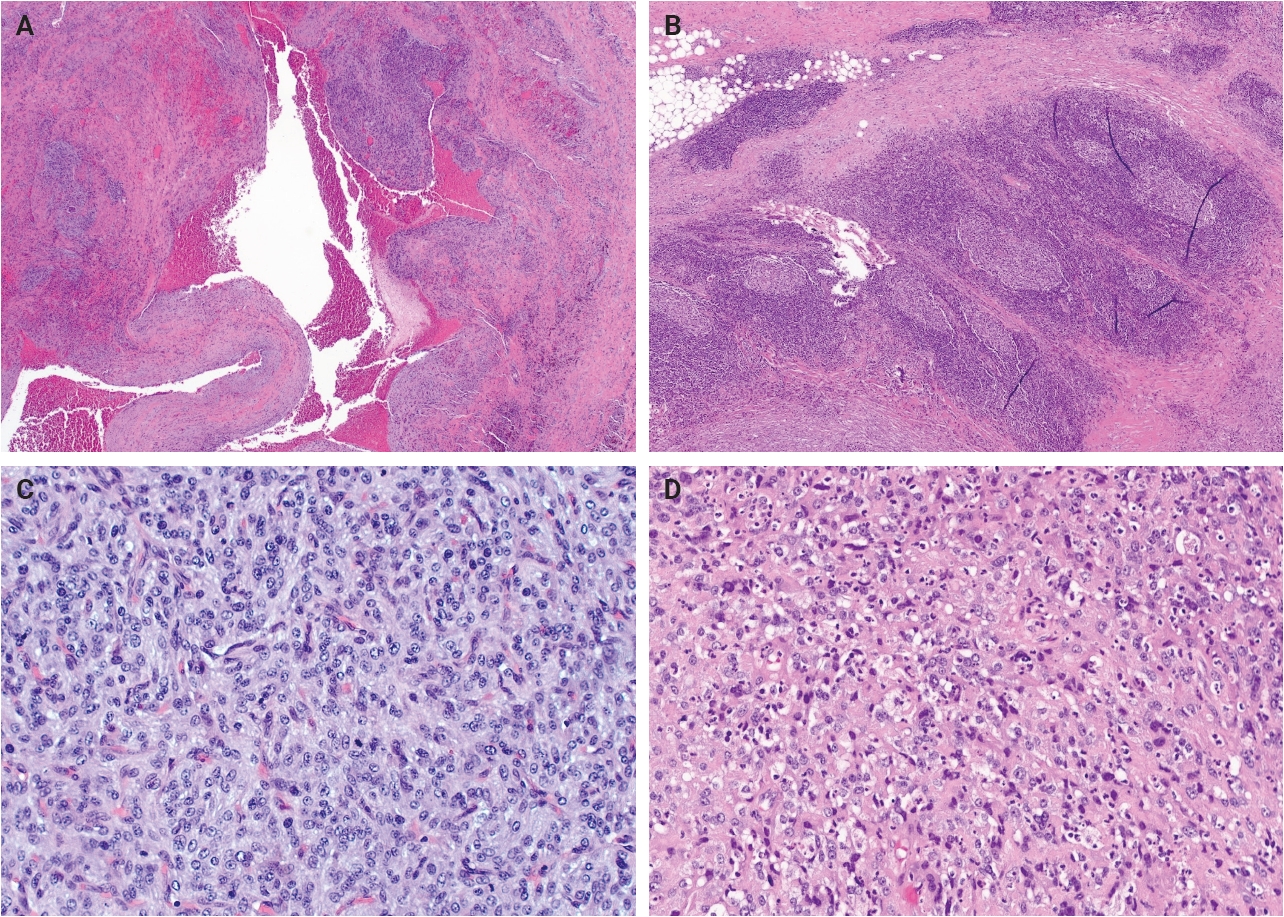
- Grossly, AFH typically presents as a well-circumscribed, firm soft tissue mass, with some cases demonstrating hemorrhagic cut surfaces. Histologically, the most characteristic and consistently observed features include sheets or fascicles of spindled-to-histiocytoid cells, central hemorrhagic or pseudocystic spaces, and a lymphoid cuff composed predominantly of chronic inflammatory cells, findings that are present in the majority, but not all, of cases (Fig. 1) [3,6,14,15]. Tumor cells are generally plump and are often arranged in syncytial sheets. At the periphery of the lesion, there is frequently a dense rim of lymphocyte-rich chronic inflammatory infiltrate with well-formed germinal centers. This feature may lead to diagnostic confusion with a lymph node or lymphoid neoplasm [1,4,6].
- Mitotic activity is generally low or absent; however, a small subset of cases demonstrates increased mitotic activity (>5 mitoses per 10 high-power fields). Some studies have suggested a possible association between elevated mitotic activity and an increased risk of local recurrence or metastasis, although a definitive prognostic correlation has not been established [6,14,15]. Additionally, focal or scattered pleomorphic tumor cells may occasionally be identified, though the biologic significance of this finding remains uncertain [14,16]. Similar to other neoplasms of intermediate biologic potential, local recurrence appears to be more strongly associated with incomplete excision than with cytologic atypia [6].
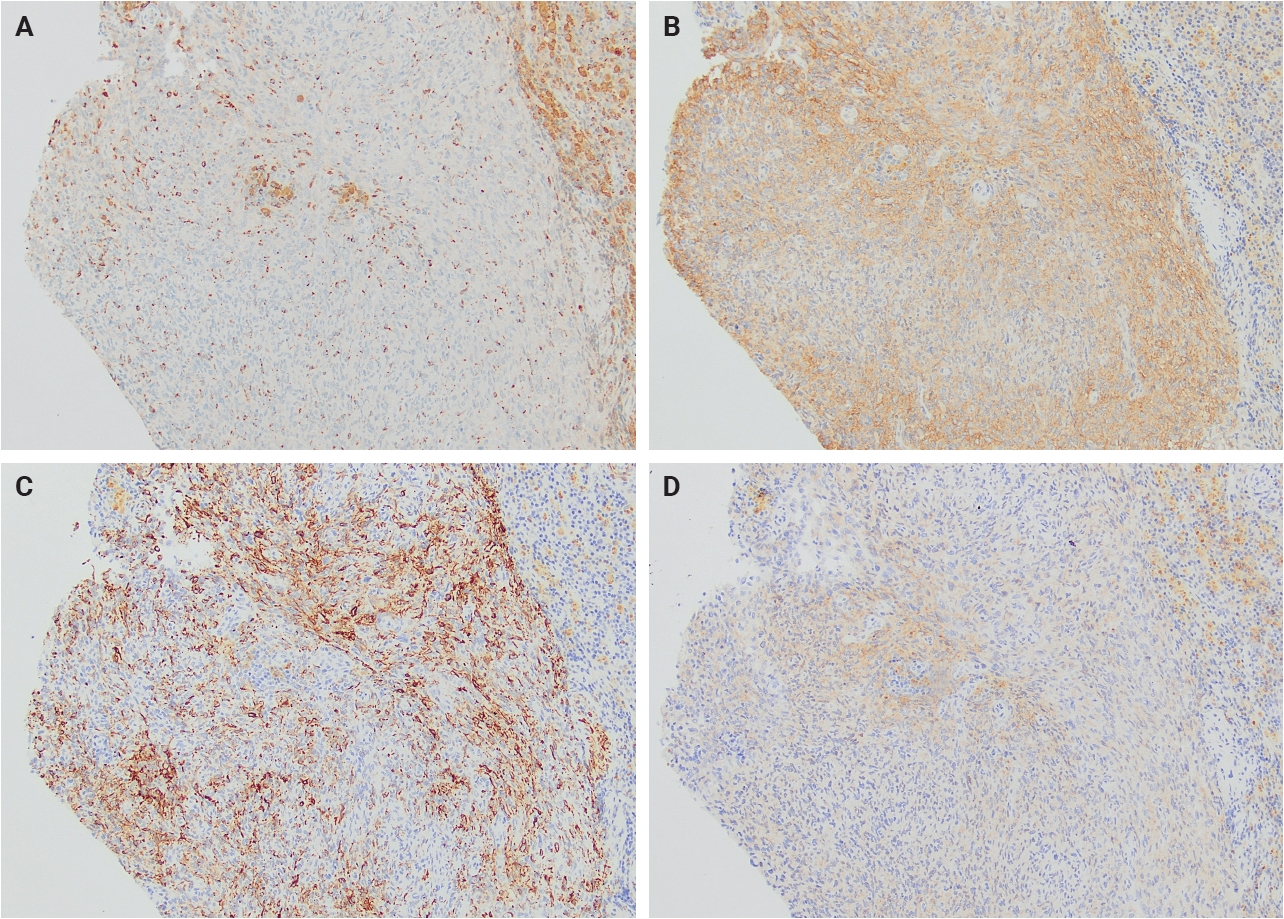
- Immunohistochemically, AFH demonstrates a relatively characteristic but nonspecific immunophenotype (Table 1). Most tumors express variable combinations of desmin, epithelial membrane antigen (EMA), CD68, CD99, and calponin, with each marker reported in approximately half of cases (Fig. 2). Despite the relatively frequent expression of desmin and calponin, there is no convincing evidence supporting true skeletal muscle differentiation, as MyoD1 and myogenin are consistently negative [4].
- Additional immunohistochemical markers have been investigated in recent years. Anaplastic lymphoma kinase (ALK) expression, particularly with the D5F3 clone, has been reported in a subset of AFH cases despite the absence of corresponding ALK gene fusions/rearrangements or copy number alterations [18-20]. Furthermore, variable MUC4 expression has been identified in a minority of tumors [21]. Awareness of these staining patterns is important to avoid potential diagnostic confusion with inflammatory myofibroblastic tumor in ALK-positive cases or with low-grade fibromyxoid sarcoma/sclerosing epithelioid fibrosarcoma in MUC4-positive lesions.
PATHOLOGIC FINDINGS
- The FET family of RNA-binding proteins, comprising EWSR1 (Ewing sarcoma breakpoint region 1), FUS (fused in sarcoma), and TAF15 (TATA-box binding protein-associated factor 15), plays a central role in the pathogenesis of numerous fusion-driven mesenchymal neoplasms. Members of this family frequently function as interchangeable fusion partners in a wide variety of sarcomas and other soft tissue tumors. Early molecular studies of AFH identified EWSR1::ATF1 and FUS::ATF1 fusion events. However, subsequent investigations have demonstrated that the most common molecular abnormality in AFH is the EWSR1::CREB1 fusion, most frequently involving exon 7 of EWSR1 fused to exon 7 of CREB1 [14,22,23]. Less commonly, alternative fusion partners have been described, including rare variant fusions such as EWSR1::PBX3, which nevertheless appear to retain morphologic features characteristic of conventional AFH [14].
- The cyclic AMP response element-binding protein (CREB) family of transcription factors is involved in the regulation of cellular proliferation, differentiation, and survival. Aberrant activation of CREB-family transcription factors, whether through fusion events, constitutive activation, or other molecular alterations, has been implicated in the pathogenesis of a broad spectrum of neoplasms, including hematologic malignancies and sarcomas [11]. In AFH, fusion-driven activation of CREB-family members is believed to contribute to tumorigenesis through dysregulated transcriptional signaling pathways.
- Given the presence of overlapping molecular alterations, particularly involving EWSR1 and CREB-family fusion partners, AFH shares molecular similarities with several other neoplasms. These relationships have prompted comparisons with a variety of tumors arising in soft tissue, visceral, and intracranial locations, which are discussed in the Differential Diagnosis section below.
MOLECULAR AND CYTOGENETICS
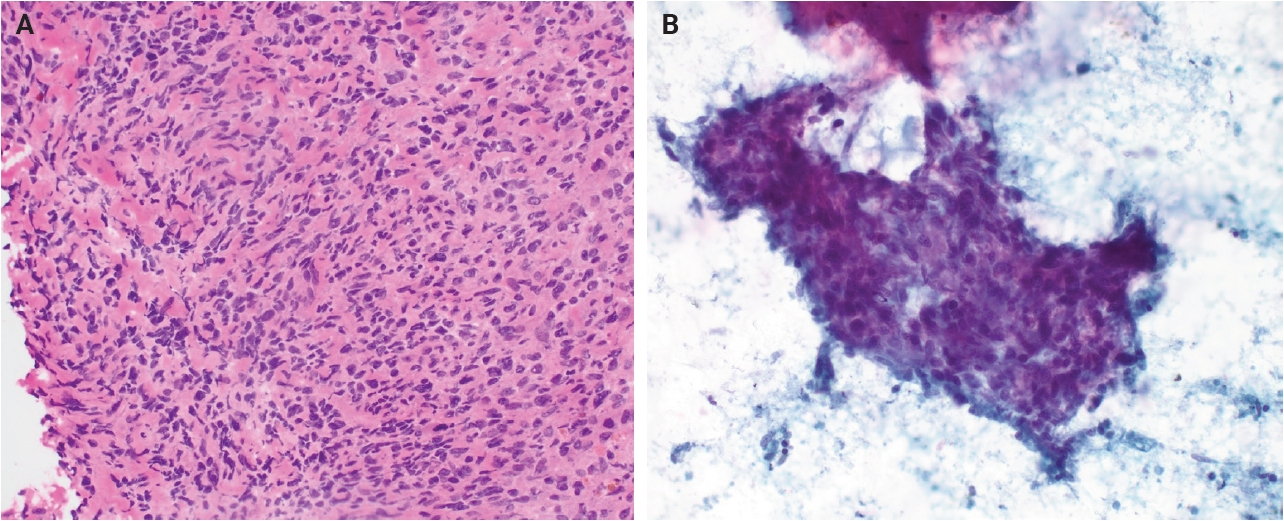
- Morphologically, the differential diagnosis for AFH includes both reactive and neoplastic entities. Reactive conditions such as non-necrotizing granulomatous lymphadenitis may enter the differential diagnosis, particularly when AFH arises in nodal or peri-nodal locations, which can confound examination by intraoperative or cytologic means (Fig. 3). However, granulomatous lesions typically contain necrotic or fibrinoid central material rather than the hemorrhagic pseudocystic spaces characteristic of AFH. Among neoplastic mimickers, follicular dendritic cell sarcoma (FDCS) may show overlapping features, including a lymphoid-rich background. Nevertheless, FDCS more commonly demonstrates uniform cytologic atypia, fascicular architecture, and inflammation distributed throughout the tumor rather than confined to the periphery, as seen in AFH. Immunohistochemically, AFH may be distinguished by expression of desmin and/or EMA and lack of follicular dendritic cell markers such as CD21, CD23, and CD35.
- Vascular neoplasms, particularly epithelioid hemangioma (formerly termed angiolymphoid hyperplasia with eosinophilia), may also be considered in cases with prominent hemorrhage. In contrast to AFH, epithelioid hemangioma demonstrates well-formed vascular channels and generally lacks the solid cellularity and syncytial growth pattern characteristic of AFH. Endothelial markers such as ERG and CD31 are helpful in excluding vascular neoplasia.
- In atypical cases, particularly those with increased cytologic atypia or EMA expression, AFH may raise concern for metastatic carcinoma of unknown primary origin. Recognition of the generally bland cytomorphology of AFH, combined with appropriate clinicopathologic correlation and exclusion of a known primary epithelial malignancy, is essential in avoiding this diagnostic pitfall. Perhaps the most important histologic mimicker of AFH is aneurysmal fibrous histiocytoma (also termed aneurysmal dermatofibroma), a variant of cellular fibrous histiocytoma. Like AFH, these lesions may exhibit extensive hemorrhagic change and hemosiderin deposition. However, aneurysmal fibrous histiocytoma typically demonstrates slender spindle cells arranged in a storiform pattern, rather than the sheet-like or syncytial architecture seen in AFH. In addition, the dense peripheral lymphoid cuff characteristic of AFH is generally absent. Clinically, aneurysmal fibrous histiocytoma behaves in an overwhelmingly indolent manner, although rare metastatic cases have been reported [24,25].
- The molecular profile of AFH, particularly its recurrent FET-CREB family gene fusions, has raised the possibility of biologic relationships with several neoplasms arising in other anatomic sites. One such entity is primary pulmonary myxoid sarcoma (PPMS), a rare pulmonary neoplasm characterized by cords, reticular growth, and myxoid stroma [26]. Some cases of PPMS exhibit morphologic overlap with AFH, including syncytial architecture and peripheral lymphoid cuffs. However, PPMS generally demonstrates more rounded or slender spindle cells within a distinctly myxoid background. Immunohistochemically, EMA expression may be seen in occasional cases, although desmin expression is typically absent. The presence of EWSR1::CREB1 fusions in most PPMS cases has led to speculation that these lesions may represent part of a related biologic spectrum [27].
- Within the central nervous system, there is a recently described “Intracranial mesenchymal tumor with FET-CREB fusion,” which shares substantial morphologic and molecular overlap with AFH. These tumors may arise intracranially or within the spinal canal and can closely resemble AFH histologically. Interestingly, a female predominance has been reported, with some studies suggesting ratios approaching 4:1. Larger series have demonstrated potential correlations between specific fusion types and morphologic patterns, and epigenetic profiling has suggested possible subclassification schemes. A minority of cases have additionally shown rearrangements involving members of the SMARCB/SMARCA family, alterations not typically identified in conventional AFH [28,29]. Given the marked overlap in morphology and cytogenetic features, these tumors are widely considered to be closely related to AFH.
- Other neoplasms harboring similar molecular alterations are generally more readily distinguished from AFH based on morphology and immunophenotype. Clear cell sarcoma, most commonly associated with the EWSR1::ATF1 fusion, demonstrates nests and fascicles of uniform spindle-to-epithelioid cells with clear cytoplasm, prominent nucleoli, and sclerotic stroma. These tumors also show diffuse melanocytic differentiation, with strong expression of S100, SOX10, and HMB45 [30]. In addition, emerging reports have described intra-abdominal malignant epithelioid neoplasms with FUS::CREM rearrangements, which are characteristically strongly and diffusely positive for cytokeratins and lack significant morphologic overlap with AFH [31].
- Other uncommon considerations include inflammatory myofibroblastic tumor, myoepithelial neoplasms with EWSR1 rearrangements, synovial sarcoma with lymphoid-rich stroma, and, rarely, CIC-rearranged sarcoma, in the appropriate clinicopathologic context.
DIFFERENTIAL DIAGNOSIS (AND POSSIBLY RELATED ENTITIES)
- Angiomatoid fibrous histiocytoma (AFH) is classified as an intermediate (rarely metastasizing) neoplasm with indeterminate biologic potential, reflecting its tendency for local recurrence but overall low metastatic risk. Accordingly, complete surgical excision remains the mainstay of treatment [32]. Local recurrence represents the most common adverse clinical outcome, although it occurs relatively infrequently, reported in fewer than 10% of cases and as low as approximately 1% in some series. Metastatic disease is distinctly uncommon but has been documented in rare instances, involving both regional lymph nodes and the lungs [4,6,24,33].
- As discussed previously, a subset of AFH cases demonstrates excess IL-6 production, resulting in paraneoplastic inflammatory symptoms. In this setting, IL-6–targeted therapies have been utilized for symptom control, with variable reported effectiveness regarding both systemic symptom improvement and tumor response [12,13,34].
- In unresectable cases or in patients for whom surgery is not feasible, systemic chemotherapy has been employed in small case series and reports, with outcomes ranging from partial response to complete radiologic remission at primary and/or metastatic sites [35]. Additionally, although ALK expression has been identified immunohistochemically in a subset of AFH cases, corresponding ALK gene rearrangements are generally absent. Nevertheless, isolated reports have suggested that ALK expression may carry therapeutic relevance, and ALK-targeted agents, similar to those used in ALK-positive non–small cell lung carcinoma, have been administered in metastatic AFH [36].
- More recently, programmed death-ligand 1 expression has been investigated in AFH, with at least focal positivity identified in a majority of tested cases (approximately 60% of reported cases). Although the clinical significance of this finding remains uncertain, it raises the possibility that immune checkpoint inhibitor therapy could represent a future therapeutic option, particularly in advanced, unresectable, or metastatic disease settings where conventional surgical management is not feasible.
TREATMENT AND PROGNOSIS
- Angiomatoid fibrous histiocytoma (AFH) is a rare mesenchymal neoplasm of uncertain differentiation that most commonly arises within the subcutaneous soft tissues of children and young adults, although it may occur across a broad age range and at diverse anatomic sites. Despite its generally indolent clinical course, AFH is classified as an intermediate (rarely metastasizing) tumor because of its recognized potential for local recurrence—particularly following incomplete excision—and its low but well-documented metastatic risk.
- Diagnosis of AFH relies on the integration of morphologic, immunohistochemical, and molecular findings. The differential diagnosis encompasses a variety of reactive and neoplastic entities, including tumors with overlapping histologic features as well as lesions sharing similar molecular alterations involving FET-CREB family fusions. In most cases, careful histopathologic evaluation combined with appropriate immunohistochemical, and molecular studies allows accurate distinction from these mimickers.
- Continued investigation into the molecular landscape and biologic relationships of AFH may further clarify its histogenesis and potential connections with other FET-CREB fusion–associated neoplasms, while also opening avenues for targeted therapeutic strategies in advanced or unresectable disease.
CONCLUSION
Ethics Statement
Not applicable.
Availability of Data and Material
Data sharing not applicable to this article as no datasets were generated or analyzed during the study.
Code Availability
Not applicable.
Author Contributions
Conceptualization: ANP, PPA. Data curation: ANP. Writing—original draft: ANP. Writing—review & editing: ANP, PPA. Approval of final manuscript: all authors.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
Funding Statement
No funding to declare.
| Marker | Frequency (%) |
|---|---|
| CD99 | 50–90 |
| Desmin | 50 to >90 |
| EMA | >70 |
| CD68 | ~50 |
| Calponin | 73 (focal) and 12 (diffuse) |
| ALK | >80 |
| MUC4 | ~20 |
Data summarized from PathologyOutlines.com (https://www.pathologyoutlines.com/topic/skintumornonmelanocyticafh.html) [17].
- 1. Enzinger FM. Angiomatoid malignant fibrous histiocytoma: a distinct fibrohistiocytic tumor of children and young adults simulating a vascular neoplasm. Cancer 1979; 44: 2147-57. ArticlePubMed
- 2. WHO Classification of Tumours Editorial Board. WHO Classification of tumours, 5th ed. Vol. 3. Soft tissue and bone tumours [Internet]. Lyon: International Agency for Research on Cancer, 2020 [cited 2026 May 8]. Available from: https://publications.iarc.fr/588.
- 3. Thway K. Angiomatoid fibrous histiocytoma: a review with recent genetic findings. Arch Pathol Lab Med 2008; 132: 273-7. ArticlePubMedPDF
- 4. Fanburg-Smith JC, Miettinen M. Angiomatoid "malignant" fibrous histiocytoma: a clinicopathologic study of 158 cases and further exploration of the myoid phenotype. Hum Pathol 1999; 30: 1336-43. ArticlePubMed
- 5. Yikilmaz A, Ngan BY, Navarro OM. Imaging of childhood angiomatoid fibrous histiocytoma with pathological correlation. Pediatr Radiol 2015; 45: 1796-802. ArticlePubMedPDF
- 6. Costa MJ, Weiss SW. Angiomatoid malignant fibrous histiocytoma: a follow-up study of 108 cases with evaluation of possible histologic predictors of outcome. Am J Surg Pathol 1990; 14: 1126-32. ArticlePubMed
- 7. Argenyi ZB, Van Rybroek JJ, Kemp JD, Soper RT. Congenital angiomatoid malignant fibrous histiocytoma: a light-microscopic, immunopathologic, and electron-microscopic study. Am J Dermatopathol 1988; 10: 59-67. ArticlePubMed
- 8. Hu M, Guo F, Xiao S, et al. Primary angiomatoid fibrous histiocytoma of the mandible with EWSR1-ATF1 fusion in an adult patient: case report and review of literature. Oral Surg Oral Med Oral Pathol Oral Radiol 2023; 136: e116-22. ArticlePubMed
- 9. Maharjan S, Satyal B, Baidya R, Joshi A, Baral P. Angiomatoid fibrous histiocytoma mimicking a lymph nodal lesion: a case report. JNMA J Nepal Med Assoc 2022; 60: 200-3. ArticlePubMedPMC
- 10. Akiyama M, Yamaoka M, Mikami-Terao Y, et al. Paraneoplastic syndrome of angiomatoid fibrous histiocytoma may be caused by EWSR1-CREB1 fusion-induced excessive interleukin-6 production. J Pediatr Hematol Oncol 2015; 37: 554-9. ArticlePubMed
- 11. Siu YT, Jin DY. CREB: a real culprit in oncogenesis. FEBS J 2007; 274: 3224-32. ArticlePubMed
- 12. Villiger PM, Cottier S, Jonczy M, Koelzer VH, Roux-Lombard P, Adler S. A simple Baker's cyst? Tocilizumab remits paraneoplastic signs and controls growth of IL-6-producing angiomatoid malignant fibrous histiocytoma. Rheumatology (Oxford) 2014; 53: 1350-2. ArticlePubMed
- 13. Sabe H, Inoue A, Nagata S, et al. Tocilizumab controls paraneoplastic inflammatory syndrome but does not suppress tumor growth of angiomatoid fibrous histiocytoma. Case Rep Oncol Med 2021; 2021: 5532258.ArticlePubMedPMCPDF
- 14. Agaimy A, Molligan J, Alruwaii FI, et al. Angiomatoid fibrous histiocytoma occurring at distal/acral extremity sites: clinicopathological and molecular study of 26 cases highlighting frequent myxoid histology and site-dependent genotypic variation. Virchows Arch 2025; 487: 587-95. ArticlePubMedPMCPDF
- 15. Zeng Q, Li JZ, Li GP, Chen YP, Song FL, Gao F. Clinical and pathological analyses of 14 cases of angiomatoid fibrous histiocytoma. Med Mol Morphol 2024; 57: 299-305. ArticlePubMedPDF
- 16. Matsumura T, Yamaguchi T, Tochigi N, Wada T, Yamashita T, Hasegawa T. Angiomatoid fibrous histiocytoma including cases with pleomorphic features analysed by fluorescence in situ hybridisation. J Clin Pathol 2010; 63: 124-8. ArticlePubMed
- 17. Perez AN, Aung PP. Skin nonmelanocytic tumor: tumors of uncertain lineage angiomatoid fibrous histiocytoma [Internet]. Bingham Farms: PathologyOutlines.com, 2026 [cited 2026 May 8]. Available from: https://www.pathologyoutlines.com/topic/skintumornonmelanocyticafh.html.
- 18. Cheah AL, Zou Y, Lanigan C, et al. ALK expression in angiomatoid fibrous histiocytoma: a potential diagnostic pitfall. Am J Surg Pathol 2019; 43: 93-101. ArticlePubMed
- 19. De Noon S, Fleming A, Singh M. Angiomatoid fibrous histiocytoma with ALK expression in an unusual location and age group. Am J Dermatopathol 2020; 42: 689-93. ArticlePubMed
- 20. Van Zwam P, Mentzel T, Flucke U. ALK expression in angiomatoid fibrous histiocytoma: confirmation of the findings of Cheah et al. Am J Surg Pathol 2019; 43: 1156.ArticlePubMed
- 21. Abrahao-Machado LF, Bacchi LM, Fernandes IL, Costa FD, Bacchi CE. MUC4 expression in angiomatoid fibrous histiocytoma. Appl Immunohistochem Mol Morphol 2020; 28: 641-5. ArticlePubMed
- 22. Thway K, Fisher C. Angiomatoid fibrous histiocytoma: the current status of pathology and genetics. Arch Pathol Lab Med 2015; 139: 674-82. ArticlePubMedPDF
- 23. Antonescu CR, Dal Cin P, Nafa K, et al. EWSR1-CREB1 is the predominant gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer 2007; 46: 1051-60. ArticlePubMed
- 24. Mankertz F, Kessler R, Rau A, Seebauer C, Ribback S, Busemann A. Pulmonary metastasising aneurysmal fibrous histiocytoma: a case report, literature review and proposal of standardised diagnostic criteria. Diseases 2023; 11: 108.ArticlePubMedPMC
- 25. Nabatanzi A, Male M, Qu XY, et al. Aneurysmal fibrous histiocytoma: clinicopathology analysis of 30 cases of a rare variant of cutaneous fibrohistiocytoma. Curr Med Sci 2019; 39: 134-7. ArticlePubMedPDF
- 26. Thway K, Nicholson AG, Lawson K, et al. Primary pulmonary myxoid sarcoma with EWSR1-CREB1 fusion: a new tumor entity. Am J Surg Pathol 2011; 35: 1722-32. ArticlePubMed
- 27. Kerper AL, Larsen BT, Folpe AL, et al. Primary pulmonary myxoid sarcoma and thoracic angiomatoid fibrous histiocytoma: two sides of the same coin? Am J Surg Pathol 2024; 48: 562-9. ArticlePubMed
- 28. Sloan EA, Chiang J, Villanueva-Meyer JE, et al. Intracranial mesenchymal tumor with FET-CREB fusion: a unifying diagnosis for the spectrum of intracranial myxoid mesenchymal tumors and angiomatoid fibrous histiocytoma-like neoplasms. Brain Pathol 2021; 31: e12918.ArticlePubMedPDF
- 29. Rajan S, Chung HJ, Wu Z, et al. Intracranial mesenchymal tumor, FET::CREB fusion-positive: an integrative analysis of 81 cases. Neuro Oncol 2026; 28: 939-51. ArticlePubMedPMC
- 30. Dashti NK, Schukow CP, Kilpatrick SE. Back to the future!: selected bone and soft tissue neoplasms with shared genetic alterations but differing morphological and immunohistochemical phenotypes. Hum Pathol 2024; 147: 129-38. ArticlePubMed
- 31. Agaimy A, Stoehr R, Otto M, et al. Intra-abdominal EWSR1/FUS-CREM-rearranged malignant epithelioid neoplasms: two cases of an emerging aggressive entity with emphasis on misleading immunophenotype. Virchows Arch 2022; 480: 481-6. ArticlePubMedPMCPDF
- 32. Mazur-Hart DJ, O'Neill BE, Pang BW, et al. Operative technique: angiomatoid fibrous histiocytoma-unique case and management. J Neurol Surg Rep 2022; 83: e110-8. ArticlePubMedPMC
- 33. Thway K, Stefanaki K, Papadakis V, Fisher C. Metastatic angiomatoid fibrous histiocytoma of the scalp, with EWSR1-CREB1 gene fusions in primary tumor and nodal metastasis. Hum Pathol 2013; 44: 289-93. ArticlePubMed
- 34. Potter SL, Quintanilla NM, Johnston DK, Naik-Mathuria B, Venkatramani R. Therapeutic response of metastatic angiomatoid fibrous histiocytoma carrying EWSR1-CREB1 fusion to the interleukin-6 receptor antibody tocilizumab. Pediatr Blood Cancer 2018; 65: e27291.ArticlePubMedPMCPDF
- 35. Corley EA, Pace E, Barnacle AM, Patel PA, Thway K, Chisholm JC. Evidence of chemoresponsiveness in unresectable metastatic angiomatoid fibrous histiocytoma. J Pediatr Hematol Oncol 2023; 45: e279-84. ArticlePubMed
- 36. Ngo C, Grinda T, Boileve A, et al. Durable response to crizotinib in metastatic angiomatoid fibrous histiocytoma with EWSR1-CREB1 fusion and ALK overexpression. Ann Oncol 2022; 33: 848-50. ArticlePubMed
REFERENCES
Figure & Data
References
Citations
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
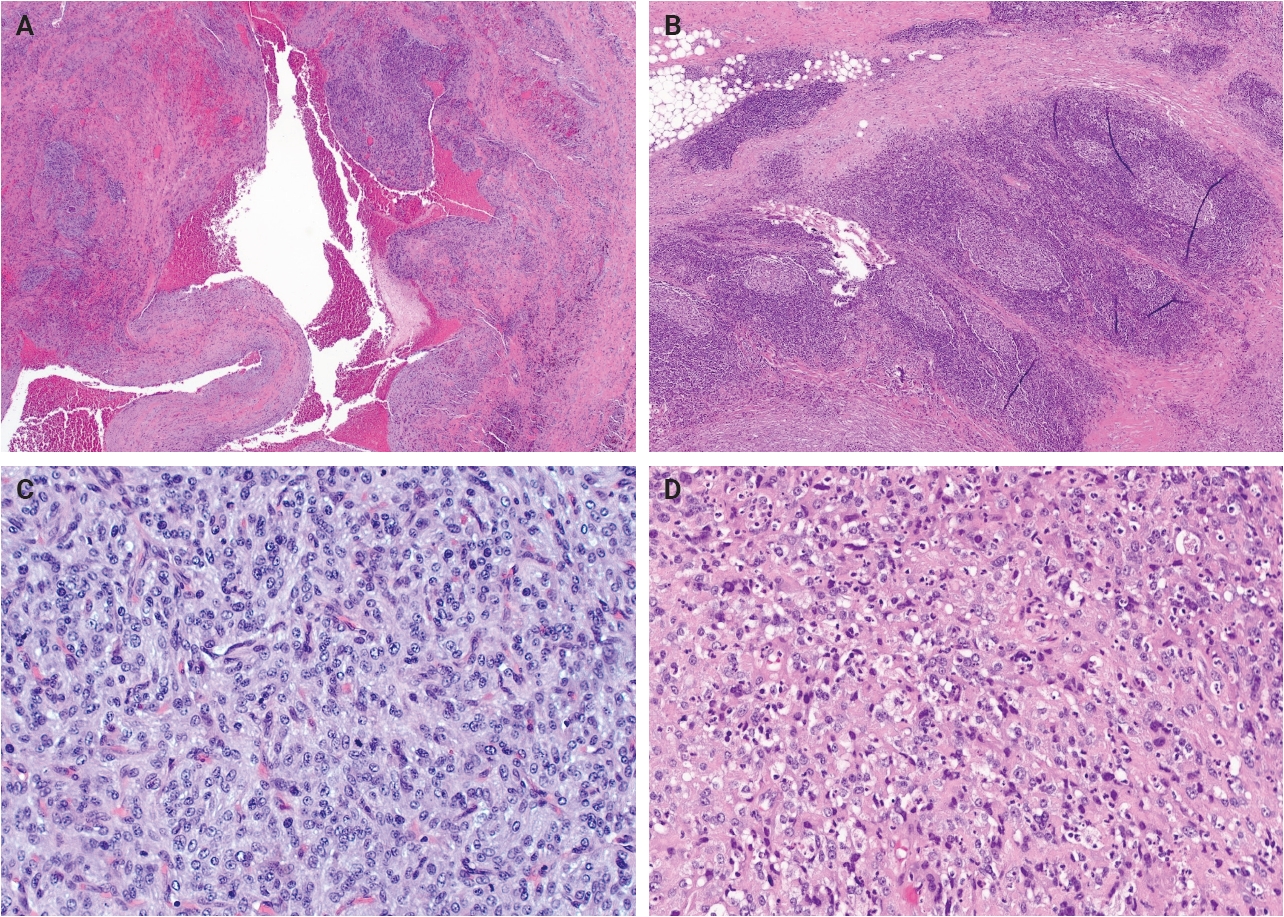
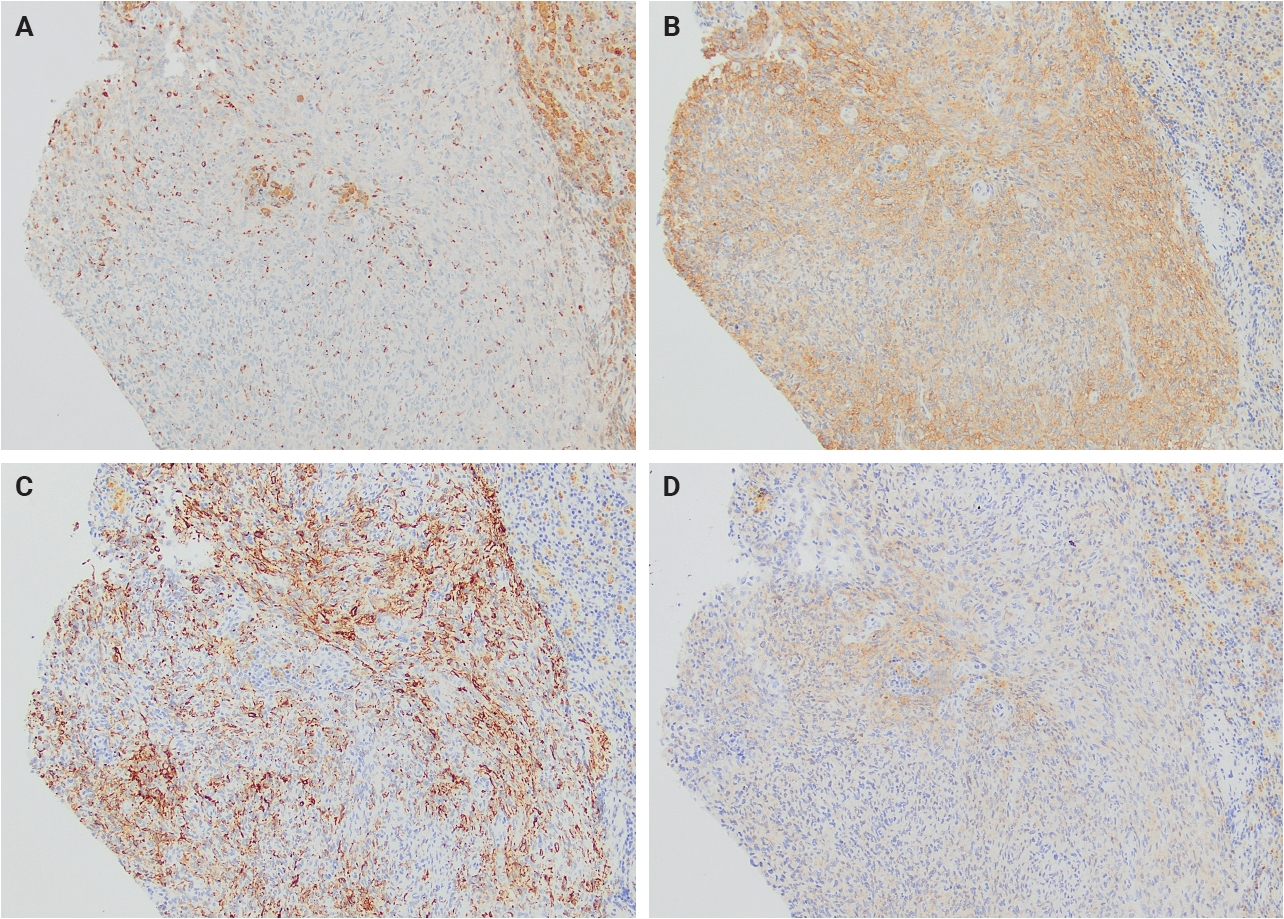
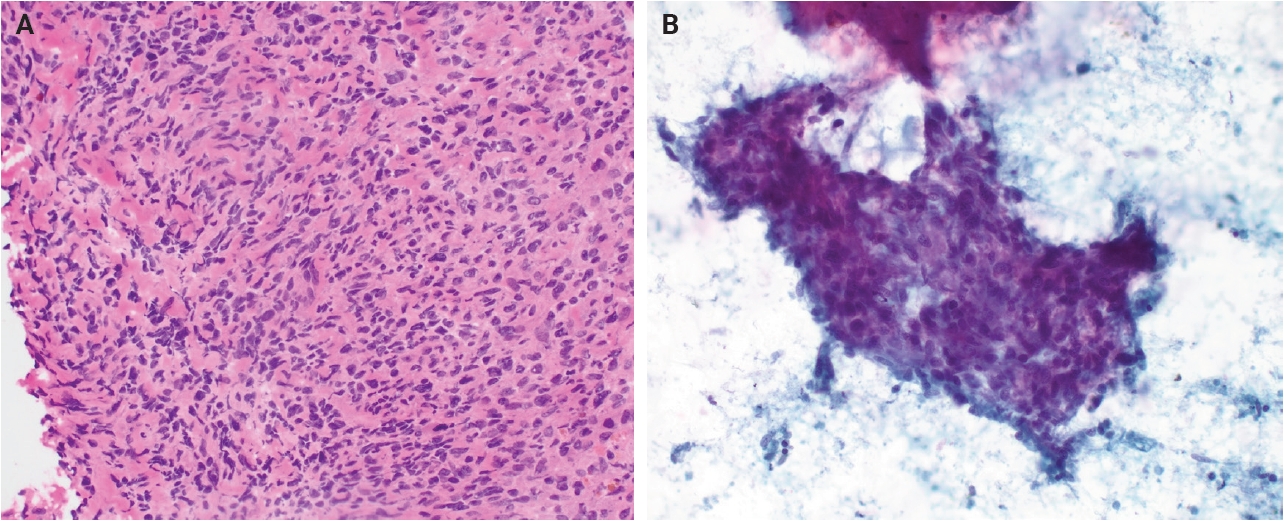

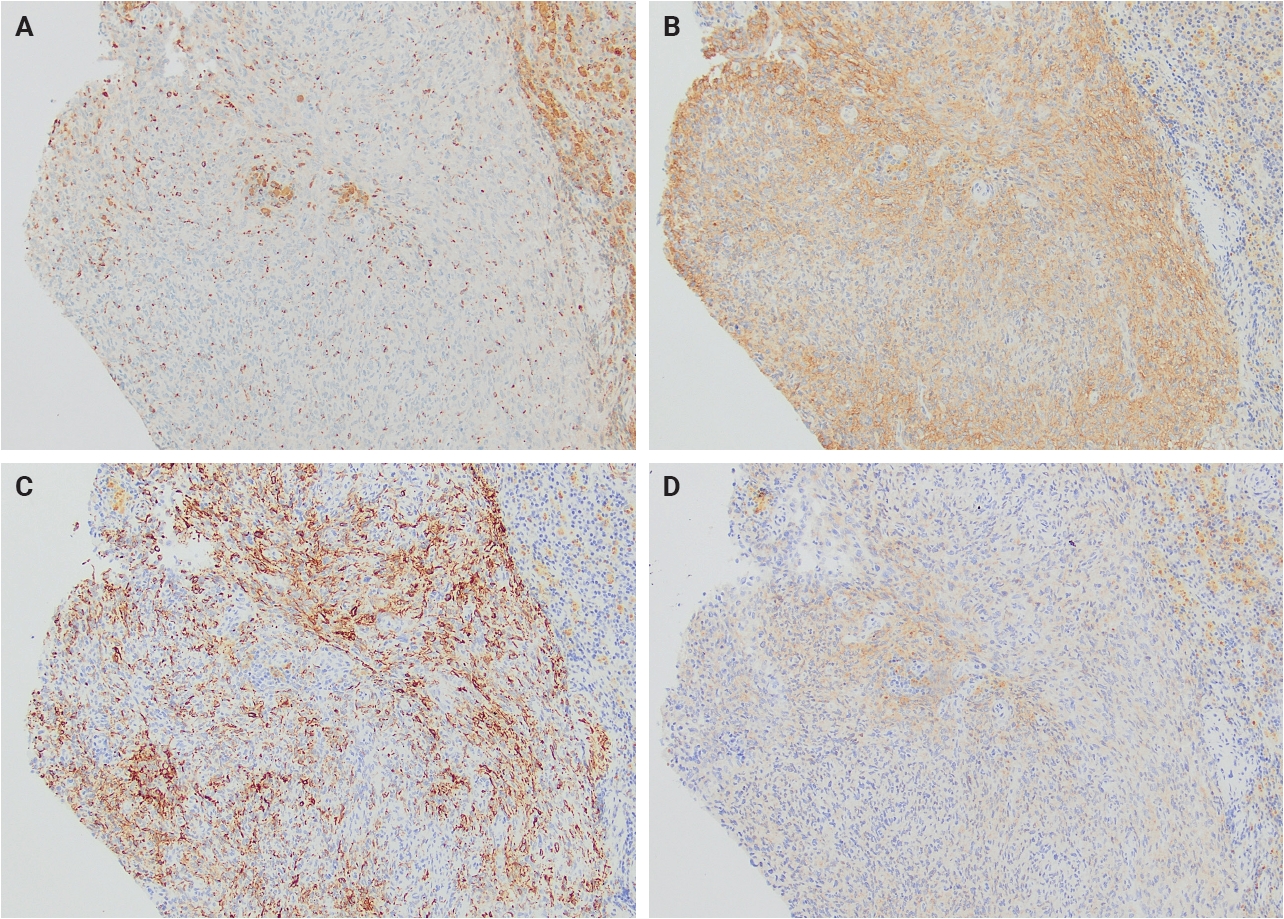
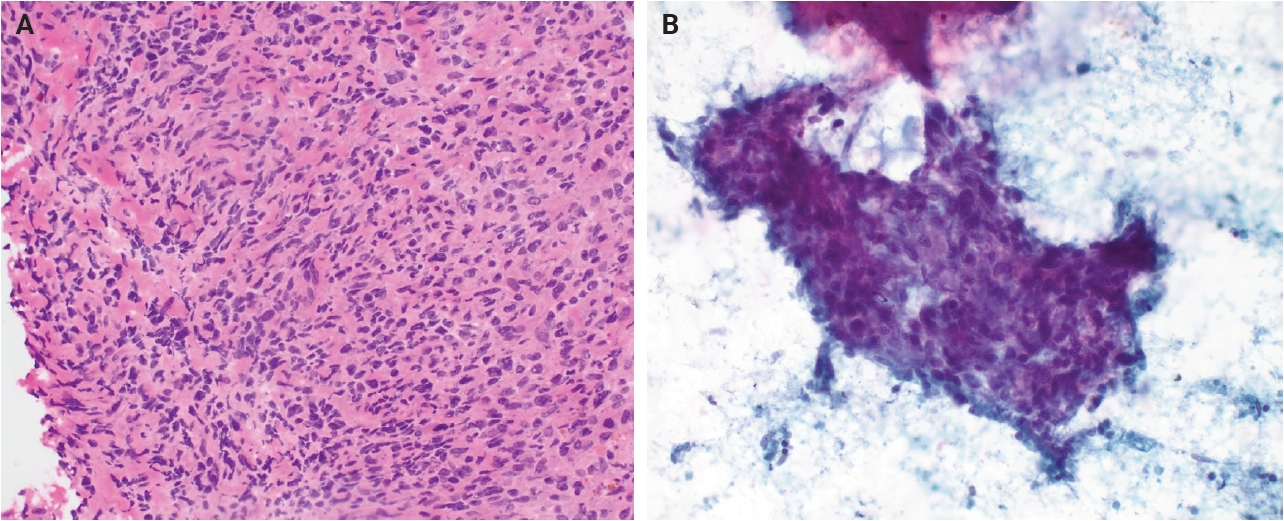
Fig. 1.
Fig. 2.
Fig. 3.
| Marker | Frequency (%) |
|---|---|
| CD99 | 50–90 |
| Desmin | 50 to >90 |
| EMA | >70 |
| CD68 | ~50 |
| Calponin | 73 (focal) and 12 (diffuse) |
| ALK | >80 |
| MUC4 | ~20 |
Data summarized from PathologyOutlines.com (

