 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 47(2); 2013 > Article
-
Original Article
EBV-Positive T/NK-Cell Lymphoproliferative Disease of Childhood - Mineui Hong, Young Hyeh Ko, Keon Hee Yoo1, Hong Hoe Koo1, Seok Jin Kim2, Won Seog Kim2, Heejung Park3
-
Korean Journal of Pathology 2013;47(2):137-147.
DOI: https://doi.org/10.4132/KoreanJPathol.2013.47.2.137
Published online: April 24, 2013
Department of Pathology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
1Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
2Department of Hematology-Oncology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
3Department of Pathology, Ewha Womans University Mokdong Hospital, Ewha Womans University School of Medicine, Seoul, Korea.
-
Corresponding Author: Young Hyeh Ko, M.D. Department of Pathology, Samsung Medical Center, Sungkyunkwan University School of Medicine, 81 Irwon-ro, Gangnam-gu, Seoul 135-710, Korea. Tel: +82-2-3410-2762, Fax: +82-2-3410-6398, yhko310@skku.edu
Corresponding Author: Heejung Park, M.D. Department of Pathology, Ewha Womans University Mokdong Hospital, Ewha Womans University School of Medicine, 1071 Anyangcheon-ro, Yangcheon-gu, Seoul 158-710, Korea. Tel: +82-2-2650-5193, Fax: +82-2-2650-2016, zealot096@gmail.com
© 2013 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
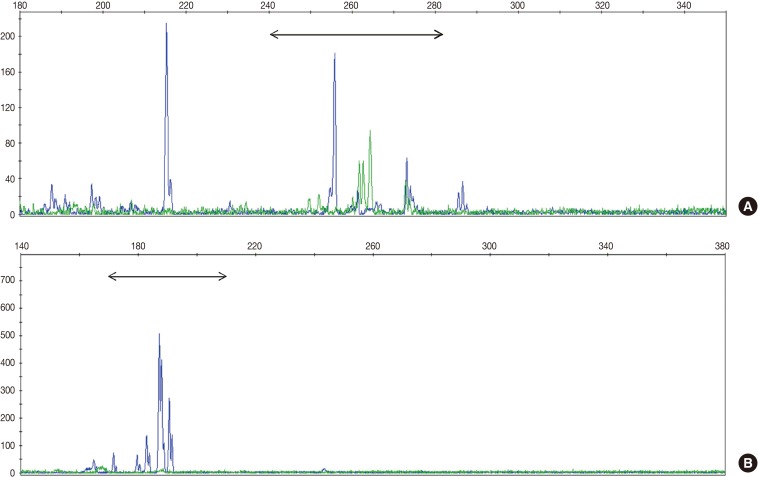

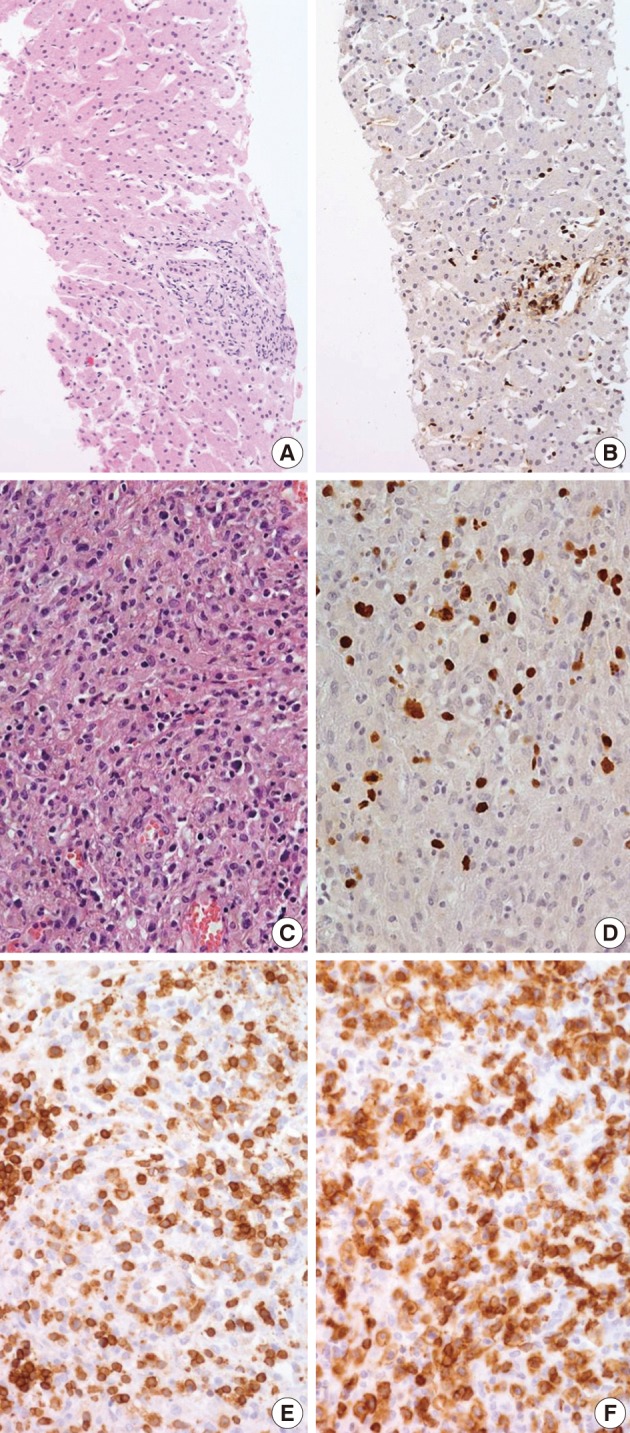
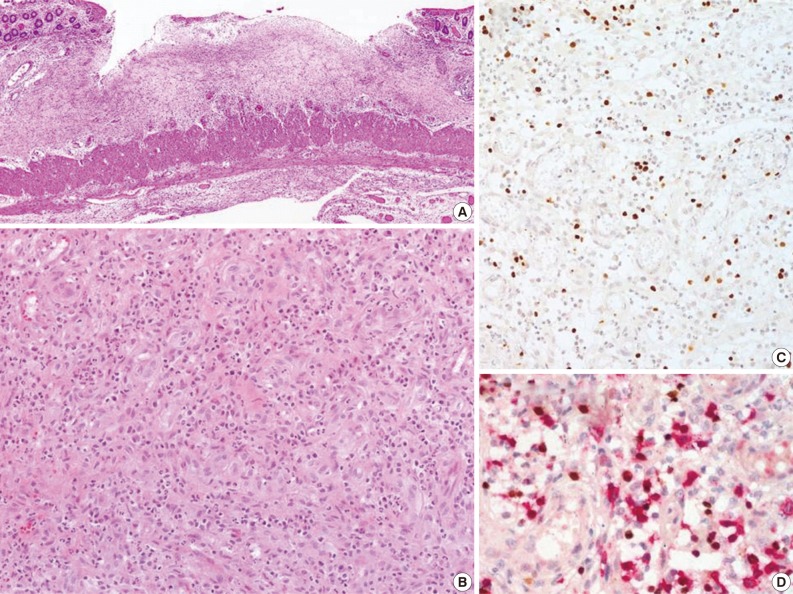
Figure & Data
References
Citations
- Chronic active Epstein-Barr virus disease: molecular pathogenesis, evolving concepts, and emerging therapies
Hiroshi Kimura, Jeffrey I. Cohen
Blood.2026; 147(14): 1562. CrossRef - Histopathological characteristics of Epstein-Barr virus (EBV)–associated encephalitis and colitis in chronic active EBV infection
Betty A Kasimo, James J Yahaya, Sun Och Yoon, Se Hoon Kim, Minsun Jung
Journal of Pathology and Translational Medicine.2025; 59(3): 188. CrossRef - Die fünfte Auflage der WHO‐Klassifikation – Was ist neu für kutane Lymphome?
Susanne Melchers, Jana D. Albrecht, Werner Kempf, Jan P. Nicolay
JDDG: Journal der Deutschen Dermatologischen Gesellschaft.2024; 22(9): 1254. CrossRef - Fifth Edition of the World Health Organization Classification of Tumors of the Hematopoietic and Lymphoid Tissues: Mature T-Cell, NK-Cell, and Stroma-Derived Neoplasms of Lymphoid Tissues
Roberto N. Miranda, Catalina Amador, John K.C. Chan, Joan Guitart, Karen L. Rech, L. Jeffrey Medeiros, Kikkeri N. Naresh
Modern Pathology.2024; 37(8): 100512. CrossRef - Clinical epidemiology of Epstein-Barr virus-associated Lymphoproliferative Disorders (EBV-LPDs) in hospitalized children: A six-year multi-institutional study in China
Dilara Dilmurat, Xinyu Wang, Liwei Gao, Jiao Tian, Junhong Ai, Linlin Zhang, Mengjia Liu, Guoshuang Feng, Yueping Zeng, Ran Wang, Zhengde Xie
Italian Journal of Pediatrics.2024;[Epub] CrossRef - The fifth edition of the WHO‐Classification – what is new for cutaneous lymphomas?
Susanne Melchers, Jana D. Albrecht, Werner Kempf, Jan P. Nicolay
JDDG: Journal der Deutschen Dermatologischen Gesellschaft.2024; 22(9): 1254. CrossRef - An update on Epstein-Barr virus–and human T-lymphotropic virus type-1–induced cutaneous manifestations. CME Part II
Alejandro A. Gru, Jose A. Plaza, Jose A. Sanches, Denis Miyashiro, Omar P. Sangueza, Francisco Bravo Puccio, Sonia Toussaint, J. Martin Sangueza
Journal of the American Academy of Dermatology.2023; 88(5): 983. CrossRef - The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms
Rita Alaggio, Catalina Amador, Ioannis Anagnostopoulos, Ayoma D. Attygalle, Iguaracyra Barreto de Oliveira Araujo, Emilio Berti, Govind Bhagat, Anita Maria Borges, Daniel Boyer, Mariarita Calaminici, Amy Chadburn, John K. C. Chan, Wah Cheuk, Wee-Joo Chng,
Leukemia.2022; 36(7): 1720. CrossRef - Chronic active Epstein–Barr virus enteritis: A literature review
Yang Shen, Yu Fang Wang
Journal of Digestive Diseases.2022; 23(5-6): 248. CrossRef - EBV-Associated Lymphoproliferative Disorders
Young Hyeh Ko
Clinical Pediatric Hematology-Oncology.2021; 28(1): 14. CrossRef - Clinicopathologic findings of chronic active Epstein–Barr virus infection in adults: A single-center retrospective study in China
Jing Lin, Haicong Wu, Lei Gu, Xia Wu, Miaofang Su, Haiyan Lin, Bang Liu, Jiaolong Zheng, Xuan Mei, Dongliang Li
Clinical and Experimental Medicine.2021; 21(3): 369. CrossRef - Outcome of L-DEP regimen for treatment of pediatric chronic active Epstein–Barr virus infection
Honghao Ma, Liping Zhang, Ang Wei, Jun Yang, Dong Wang, Qing Zhang, Yunze Zhao, Sitong Chen, Hongyun Lian, Li Zhang, Chunju Zhou, Maoquan Qin, Zhigang Li, Tianyou Wang, Rui Zhang
Orphanet Journal of Rare Diseases.2021;[Epub] CrossRef - Epstein-Barr virus NK and T cell lymphoproliferative disease: report of a 2018 international meeting
Jeffrey I. Cohen, Keiji Iwatsuki, Young-Hyeh Ko, Hiroshi Kimura, Irini Manoli, Koichi Ohshima, Stefania Pittaluga, Leticia Quintanilla-Martinez, Elaine S. Jaffe
Leukemia & Lymphoma.2020; 61(4): 808. CrossRef - EBV-positive T/NK-associated lymphoproliferative disorders of childhood: A complete autopsy report
JonathanY Keow, WilliamM Stecho, AaronR Haig, NikhilA Sangle
Indian Journal of Pathology and Microbiology.2020; 63(1): 78. CrossRef - Chronic active Epstein‐Barr virus infection: A heterogeneous entity requiring a high index of suspicion for diagnosis
Sarah L. Ondrejka, Eric D. Hsi
International Journal of Laboratory Hematology.2020; 42(S1): 99. CrossRef - Epstein-Barr Virus-Associated T and NK-Cell Lymphoproliferative Diseases
Wook Youn Kim, Ivonne A. Montes-Mojarro, Falko Fend, Leticia Quintanilla-Martinez
Frontiers in Pediatrics.2019;[Epub] CrossRef - A clinicopathologic study of the spectrum of systemic forms of EBV‐associated T‐cell lymphoproliferative disorders of childhood: A single tertiary care pediatric institution experience in North America
Amy M. Coffey, Annisa Lewis, Andrea N. Marcogliese, M. Tarek Elghetany, Jyotinder N. Punia, Chung‐Che Chang, Carl E. Allen, Kenneth L. McClain, Amos S. Gaikwad, Nader Kim El‐Mallawany, Choladda V. Curry
Pediatric Blood & Cancer.2019;[Epub] CrossRef - Unusual lymphoid malignancy and treatment response in two children with Down syndrome
Ashley Geerlinks, Jennifer Keis, Bo Ngan, Amer Shammas, Reza Vali, Johann Hitzler
Pediatric Blood & Cancer.2019;[Epub] CrossRef - EBV-Positive Lymphoproliferations of B- T- and NK-Cell Derivation in Non-Immunocompromised Hosts
Stefan Dojcinov, Falko Fend, Leticia Quintanilla-Martinez
Pathogens.2018; 7(1): 28. CrossRef - Cutaneous Hematolymphoid and Histiocytic Proliferations in Children
Alejandro A Gru, Louis P Dehner
Pediatric and Developmental Pathology.2018; 21(2): 208. CrossRef - Clinicopathological categorization of Epstein–Barr virus-positive T/NK-cell lymphoproliferative disease: an analysis of 42 cases with an emphasis on prognostic implications
Jin Ho Paik, Ji-Young Choe, Hyojin Kim, Jeong-Ok Lee, Hyoung Jin Kang, Hee Young Shin, Dong Soon Lee, Dae Seog Heo, Chul-Woo Kim, Kwang-Hyun Cho, Tae Min Kim, Yoon Kyung Jeon
Leukemia & Lymphoma.2017; 58(1): 53. CrossRef - Cutaneous EBV-related lymphoproliferative disorders
Alejandro A. Gru, Elaine S. Jaffe
Seminars in Diagnostic Pathology.2017; 34(1): 60. CrossRef - T- and NK-Cell Lymphomas and Systemic Lymphoproliferative Disorders and the Immunodeficiency Setting
Dita Gratzinger, Daphne de Jong, Elaine S. Jaffe, Amy Chadburn, John K. C. Chan, John R. Goodlad, Jonathan Said, Yasodha Natkunam
American Journal of Clinical Pathology.2017; 147(2): 188. CrossRef - Systemic Epstein-Barr Virus-positive T-Cell Lymphoproliferative Disease of Childhood With Good Response to Steroid Therapy
Do-Hoon Kim, Myungshin Kim, Yonggoo Kim, Kyungja Han, Eunhee Han, Jae Wook Lee, Nack-Gyun Chung, Bin Cho
Journal of Pediatric Hematology/Oncology.2017; 39(8): e497. CrossRef - Recent advances in the risk factors, diagnosis and management of Epstein-Barr virus post-transplant lymphoproliferative disease
Paibel Aguayo-Hiraldo, Reuben Arasaratnam, Rayne H. Rouce
Boletín Médico del Hospital Infantil de México.2016; 73(1): 31. CrossRef - Severe Epstein–Barr virus infection in primary immunodeficiency and the normal host
Austen J. J. Worth, Charlotte J. Houldcroft, Claire Booth
British Journal of Haematology.2016; 175(4): 559. CrossRef - Recent advances in the risk factors, diagnosis and management of Epstein-Barr virus post-transplant lymphoproliferative disease
Paibel Aguayo-Hiraldo, Reuben Arasaratnam, Rayne H. Rouce
Boletín Médico Del Hospital Infantil de México (English Edition).2016; 73(1): 31. CrossRef - Epstein-Barr Virus–Associated Lymphomas
Ewelina Grywalska, Jacek Rolinski
Seminars in Oncology.2015; 42(2): 291. CrossRef - Epstein–Barr virus-associated T/natural killer-cell lymphoproliferative disorder in children and young adults has similar molecular signature to extranodal nasal natural killer/T-cell lymphoma but shows distinctive stem cell-like phenotype
Siok-Bian Ng, Koichi Ohshima, Viknesvaran Selvarajan, Gaofeng Huang, Shoa-Nian Choo, Hiroaki Miyoshi, Norio Shimizu, Renji Reghunathan, Hsin-Chieh Chua, Allen Eng-Juh Yeoh, Thuan-Chong Quah, Liang-Piu Koh, Poh-Lin Tan, Wee-Joo Chng
Leukemia & Lymphoma.2015; 56(8): 2408. CrossRef - An uncommon presentation of EBV-driven HLH. Primary or secondary? An ongoing dilemma
Tânia Serrão, Alexandra Dias, Pedro Nunes, António Figueiredo
BMJ Case Reports.2015; 2015: bcr2015209615. CrossRef - Hemophagocytic syndromes — An update
Gritta E. Janka, Kai Lehmberg
Blood Reviews.2014; 28(4): 135. CrossRef - Epstein–Barr virus‐associated T/natural killer‐cell lymphoproliferative disorders
Sanghui Park, Young H. Ko
The Journal of Dermatology.2014; 41(1): 29. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
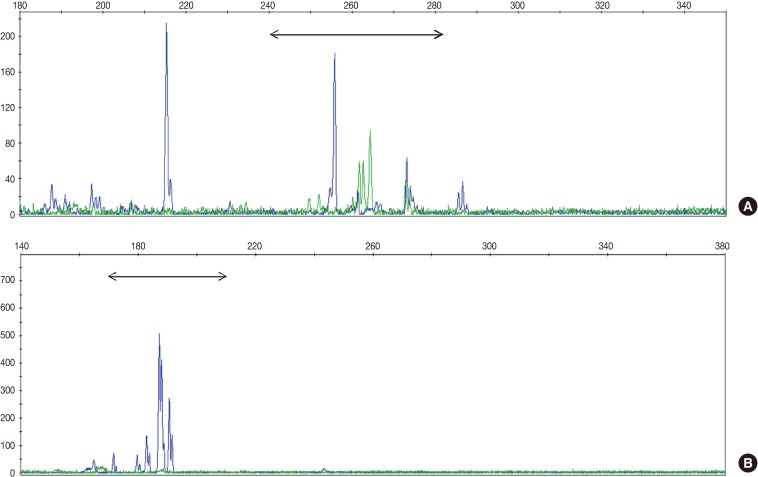
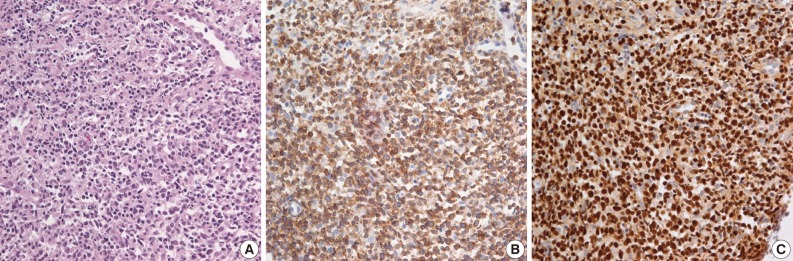
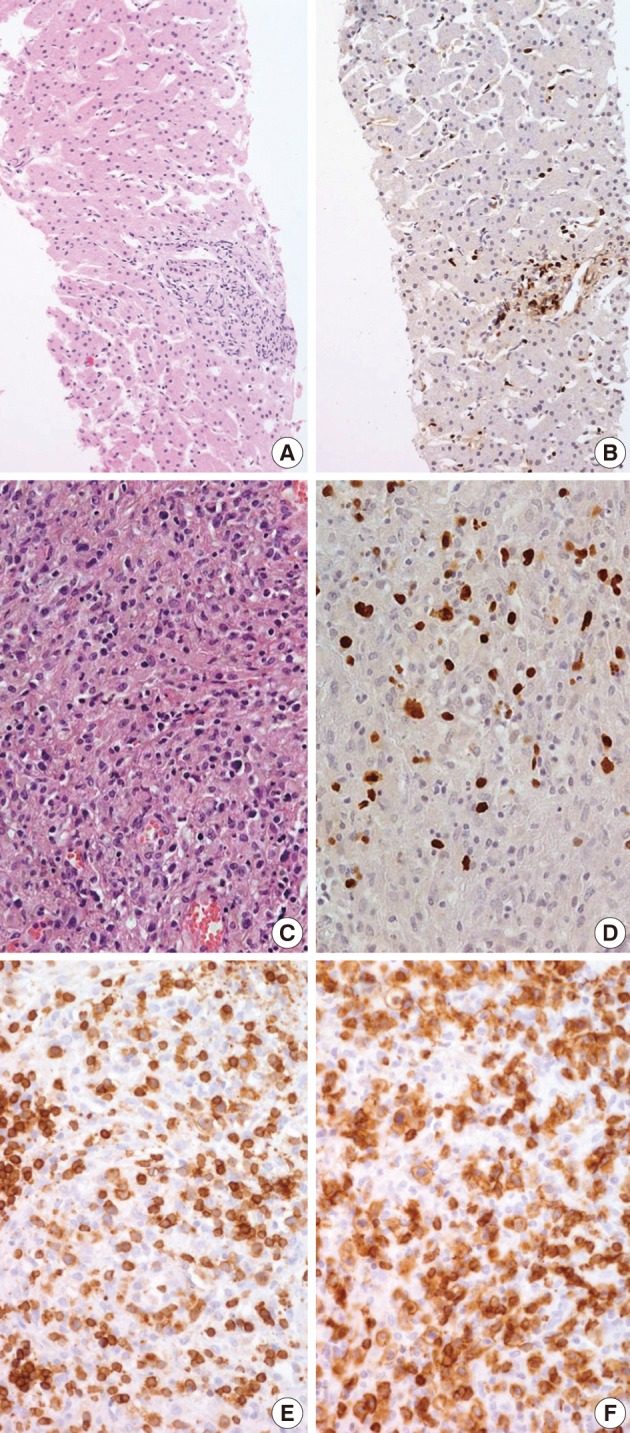
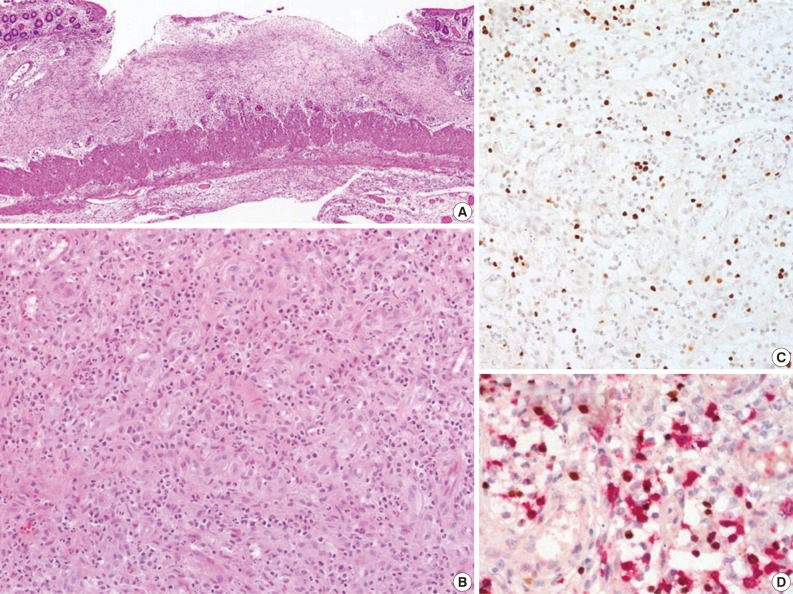




Fig. 1
Fig. 2
Fig. 3
Fig. 4
| No. | Sex | Age (yr) | Symptom | On-set | Biopsy site | Hemophagocytic histiocytosis | Cell size/Atypia | EBV-PCR (copy/μL whole blood) | EBV-ISH | EBV serology | IHC | TCRy gene | Treatment | Follow-up | Course |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Systemic T-cell LPD | |||||||||||||||
| 1 | F | 7 | Fever, cervical lymph node enlargement | 2MA | LN,BM | Present | Medium/moderate atypia | 42 (initial), 2.5-42 | Positive | EB-VCA, lgG(+) EB- | CD3+ | Monoclonal | VHR, L-Asp | 3 mo | Dead |
| VCA, IgM(-) | CD4<CD8 | ||||||||||||||
| EBV-EA(+) | CD56- | ||||||||||||||
| EBNA(+) | |||||||||||||||
| 2 | M | 43 | Fever, splenomegaly | 2MA | BM | Present | Small/mild atypia | 142 (initial), 0-142 | Positive | NC | CD3+ | Monoclonal | IMVP-16/PD | 2 mo | Dead |
| CD4+ | |||||||||||||||
| CD8+ | |||||||||||||||
| CD56– | |||||||||||||||
| EBV-positive hemophagocytic lymphohistiocytosis | |||||||||||||||
| 3 | F | 4 | Fever, jaundice, hepatomegaly, leukocytopenia | 2WA | LN, BM, liver | Present | Medium/severe atypia | 506 (initial), 4.77-6,422 | Positive | EB-VCA, igG(+) | CD3+ | Polyclonal | 106B, VHR | 3 mo | Dead |
| EB-VCA, lgM(+) | CD4- | ||||||||||||||
| EBV-EA(+) | CD8+ | ||||||||||||||
| EBNA(-) | CD56– | ||||||||||||||
| 4 | M | 10 | Cervical lymph node enlargement, persistent fever, hepatosplenomegaly, LFT abnormality, pancytopenia, facial petechiae, gingival swelling | 1MA | BM | Present | Medium/moderate atypia | 5,810 (initial), 8.4-5, 810 | Failed | NA | CD3+ | No band | HLH-2004 |
69 mo | Alive |
| CD4- | |||||||||||||||
| CD8+ | |||||||||||||||
| CD56- | |||||||||||||||
| 5 | F | 20 | Headache, fever, night sweat, abdominal pain, hepatosplenomegaly | 3DA | BM | Present | Small/mild atypia | 8.5 | Positive | NA | CD3+ | Polyclonal | CHOP | 1 mo | Dead |
| CD4<CD8 | |||||||||||||||
| CD56– | |||||||||||||||
| 6 | M | 33 | Fever | 1MA | Liver | Present | Small/mild atypia | 105.7 | Positive | NA | CD3+ | Polyclonal | Steroid | 3 days | Dead |
| CD4+CD8+ | |||||||||||||||
| CD56- | |||||||||||||||
| 7 | F | 61 | Fever, chill | 4WA | Spleen, BM | Present | Small/mild atypia | NA | Positive | EB-VCA, igG(+) | CD3+ | Polyclonal | Steroid | 1 mo | Dead |
| EB-VCA, lgM(-) | CD4+ | ||||||||||||||
| EBV-EA(-) | CDS- | ||||||||||||||
| EBNA(+) | CD56– | ||||||||||||||
| No. | Sex | Age (yr) | Symptom | On-set | Biopsy site | Hemophagocytic histiocytosis | Associated lymphoma | EBV-PCR (copy/μL whole blood) | EBV-ISH | EBV serology | IHC | TCRy gene | Treatment | Follow-up | Course |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 10 | LFT abnormality, hepatosplenomeg-aly, multiple enlarged lymph node (posterior neck, axilla and inguinal area) mosquito-bite hypersentivity | 2YA | LN, BM | Absent | Peripheral T-cell lymphoma | 77.6 (initial), 71.24-2,936 | Positive | EB-VCA, IgG(+) | CD3+ | Monoclonal/monomorphic | 106B | 10 mo | Dead |
| EB-VCA, IgM(–) | |||||||||||||||
| EBV-EA(+) | |||||||||||||||
| EBNA(+) | |||||||||||||||
| 2 | F | 14 | Fever, sore throat, IgA nephropathy, hydroa vacciniforme | Infancy | Skin, BM | Absent | T/NKcell lymphoma | NA | NA | NA | CD3+ | NA | CHOP, ESHAP | 18 mo | Dead |
| 3 | M | 15 | Nausea, weight loss, LFT abnormality, Herpes zoster infection | 2MA | Liver, BM | Present | None | 258.5 (initial), 22-258.5 | Positive | EB-VCA, IgG(+) | CD3+ | Monoclonal/monomorphic | HLH-2004 |
7 mo | Dead |
| EB-VCA, IgM(–) | CD4>CD8 | ||||||||||||||
| EBV-EA(±) | CD56– | ||||||||||||||
| EBNA(+) | |||||||||||||||
| 4 | M | 15 | Mosquito-bite hypersensitivity, palpable neck mass, NK lymphocytosis | 2WA | LN,BM | Absent | None | 529.8 (initial), 40-529.8 | Positive | EB-VCA, IgG(+) | CD3+ | NA | ABVD | 45 mo | Alive |
| EB-VCA, IgM(–) | CD4+ | ||||||||||||||
| EBV-EA(+) | CD8+ | ||||||||||||||
| EBNA(+) | CD56– | ||||||||||||||
| 5 | M | 16 | Fever and skin lesion (4YA), bowel perforation (7YA) mosquito-bite hypersensitivity, NK lymphocytosis | Infancy | Skin, BM | Present | None | 2,290 (initial), 7-2,290 | Positive | EB-VCA, IgG(+) | CD3+ | Polyclonal/polymorphic | HLH-2004 |
46 mo | Alive |
| EB-VCA, IgM(–) | CD4+ | ||||||||||||||
| EBV-EA(+) | CD8+ | ||||||||||||||
| EBNA(+) | CD56– | ||||||||||||||
| 6 | M | 21 | Mosquito-bite hypersensitivity, fever, epigastric pain, nausea, NK lymphocytosis | 7MA | Liver, BM | Present | None | NA | NA | EB-VCA, IgG(+) | NC | NA | IMVP-16PD | 3 mo | Dead |
| EB-VCA, IgM(–) | |||||||||||||||
| EBV-EA(±) | |||||||||||||||
| EBNA(+) | |||||||||||||||
| 7 | M | 21 | Chorea movement, hepatosplenomegaly, severe oral ulcer, history of pneumonia, thrombocytopenia, NK lymphocytosis | 2YA | Liver, BM | Present | None | 29 (initial), 29-73.5 | Positive | EB-VCA, IgG(+) | CD3+ | Polyclonal (KIR: NK cells with clonality) | CHOP | 10 mo | Alive (improved) |
| EB-VCA, IgM(–) | CD4>CD8 | ICE | |||||||||||||
| EBV-EA(–) | CD56+ | ||||||||||||||
| EBNA(+) | |||||||||||||||
| 8 | F | 29 | Fever, dizziness, nausea, hepatosplenomegaly, pancytopenia, LFT abnormality, diffuse lung infiltration | 2WA | LN, lung, BM | Absent | None | 189.2 (initial), 189.2-1,897 | Positive | EB-VCA, IgG(+) | CD3+ | Monoclonal/polymorphic | Self-limited | 6 mo | Alive (persistent) |
| EB-VCA, IgM(±) | CD4+ | ||||||||||||||
| EBV-EA(±) | CD8– | ||||||||||||||
| EBNA(+) | CD56– | ||||||||||||||
| 9 | M | 33 | Fatigue, hepatosplenomegaly, NK lymphocytosis | 7MA | Liver, BM | Present | None | 3,446 (initial), 1,918.6-12,112 | Positive | NA | CD3+ | NA | Refuse | 8 mo | Dead |
| CD4<CD8 | |||||||||||||||
| CD56– | |||||||||||||||
| 10 | F | 41 | Fever, hepatomegaly, abdominal pain, cerebral infact | 2YA | BM | Present | Medium/ mild atypia | 1,231.8 (initial), 1,231.8-16,188 | Positive | NA | CD3+ | Polyclonal/polymorphic | HLH-94 |
30 mo | Dead |
| CD4– | CVP | ||||||||||||||
| CD8+ | |||||||||||||||
| CD56– | |||||||||||||||
| 11 | M | 44 | LFT abnormality, hepatosplenomegaly, palpable neck mass | 8MA | LN, liver | Absent | None | NA | Positive | EB-VCA, IgG(+) | CD3+ | NA | Refuse | 6 mo | Dead |
| EB-VCA, IgM(-) | CD4+ | ||||||||||||||
| EBV-EA(+) | CD8+ | ||||||||||||||
| EBNA(+) | CD56– | ||||||||||||||
| 12 | F | 59 | Fever, myalgia, NK lymphocytosis | 7MA | BM | Present | None | 65.28 (initial), 36.6-2,934 | Negative | NA | CD3+ | No band | CVP, CHOP, IMVP-16/PD, VP | 4 mo | Dead |
| CD4– | |||||||||||||||
| CD8+ | |||||||||||||||
| CD56+ |
Treatment regimen; VHR: prednisolone, cyclophosphamide, daunorubicin, vincristine, L-asparaginase, intrathecal methotrexate, L-Asp: L-asparaginase, IMVP-16/PD: ifosfamide, methotrexate, etoposide, prednisolone, 106B: prednisolone, cyclophosphamide, daunorubicin, vincristine, L-asparaginase, CHOP: cyclophosphamide, doxorubicin, vincristine, prednisolone. LPD, lymphoproliferative disease; EBV, Epstein-Barr virus; PCR, polymerase chain reaction; ISH, HLH-94/2004: dexamethasone, cyclosporinA, intravenous Ig.
Treatment regimen; 106B: prednisolone, cyclophosphamide, daunorubicin, vincristine, L-asparaginase, CHOP: cyclophosphamide, daunorubicin, vincristine, prednisolone, ESHAP: etoposide, methylprednisolone, high-dose cytarabine, cisplatin, ABVD: adriamycin, bleomycin, vinblastine, dacarbazine, IMVP-16/PD: ifosfamide, methotrexate, etoposide, prednisolone, ICE: ifosfamide, carboplatin, etoposide, CVP: cyclophosphamide, vincristine, prednisolone. CAEBV, chronic active Epstein-Barr virus; EBV, Epstein-Barr virus; PCR, polymerase chain reaction; ISH, HLH-94/2004: dexamethasone, cyclosporinA, intravenous Ig.

