 E-submission
E-submission
Search
- Page Path
- HOME > Search
- Juxtacortical chondromyxoid fibroma in the small bones: two cases with unusual location and a literature review
- Sun-Ju Oh, So Hak Chung
- J Pathol Transl Med. 2022;56(3):157-160. Published online January 21, 2022
- DOI: https://doi.org/10.4132/jptm.2021.12.15

- 7,280 View
- 199 Download
- 3 Web of Science
- 1 Crossref
-
 Abstract
Abstract
 PDF
PDF - Chondromyxoid fibroma is a rare bone tumor of cartilaginous origin, representing less than 1% of all bone tumors. It preferentially arises in the eccentric location of the metaphysis of a long tubular bone. Juxtacortical locations are reported infrequently in the long bones and even more rarely in short tubular bones, with only three cases documented. Here we present two new cases of juxtacortical chondromyxoid fibroma in the small bones. One was an intracortical osteolytic lesion of the metatarsal bone of the foot with degenerative atypia that histologically should be differentiated from chondrosarcoma. The other was a phalangeal mass protruding into the interphalangeal joint of the hand, which had been labeled mistakenly as a soft tissue mass preoperatively. These cases illustrated that chondromyxoid fibromas have various the manifestations and should be included in the differential diagnosis of an osteolytic lesion or an exophytic mass in the small bones.
-
Citations
Citations to this article as recorded by
- Cartilage Forming Tumors of the Skeleton
Julio A. Diaz-Perez, Andrew E. Rosenberg
Advances in Anatomic Pathology.2025; 32(2): 132. CrossRef
- Cartilage Forming Tumors of the Skeleton
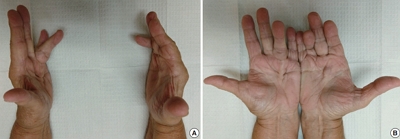
- Palmar and plantar fibromatosis: a review
- Brian D. Stewart, Alessandra F. Nascimento
- J Pathol Transl Med. 2021;55(4):265-270. Published online July 7, 2021
- DOI: https://doi.org/10.4132/jptm.2021.06.14
- 21,497 View
- 448 Download
- 19 Web of Science
- 16 Crossref
-
Abstract
PDF
- Palmar fibromatosis (Dupuytren disease/contracture) is the most common type of fibromatosis, defined as a benign proliferation of fibroblasts and myofibroblasts. The disease process is most common in white, middle-aged and older men occurring at the distal palmar crease leading to nodules and contracture, which in many cases recur after surgical treatment. In a similar process, plantar fibromatosis (Ledderhose disease) is a proliferation of fibroblasts and myofibroblasts on the plantar aponeurosis of mostly middle-aged patients that may lead to painful nodules but usually does not lead to contracture. Both processes are histologically similar, composed of a bland cellular proliferation of spindle cells with a bluish appearance and with a variable amount of background collagen, depending on the age of the lesion. The etiology of both lesions is still uncertain, while treatment ranges from observation to surgery, with some pharmacologic agents being investigated with mixed success. In this paper we provide an overview of both processes with regards to clinical and radiologic findings, pathophysiology, diagnosis, treatment, and prognosis.
-
Citations
Citations to this article as recorded by- APC-Mutated MUC4-Positive Fibroblastoma With Cytoplasmic β-Catenin: A Novel Variant Expanding Its Immunophenotypic Spectrum
Amanda Onalaja-Underwood, Rana Naous
Cureus.2026;[Epub] CrossRef - Ultrasonography of benign pediatric fibrous and adipocytic lumps
Georgios A Sideris, Madeline Stever, Mansha Khubchandani, Ziyu Xian, Michael J Callahan, Joseph Makris
Pediatric Radiology.2026; 56(7): 1429. CrossRef - Soleful solutions: Advancements in treatment strategies for ledderhose disease
Jamie Tersago, Alina Constantin
Foot and Ankle Surgery.2025; 31(1): 10. CrossRef - Emphasis on the management of embedded capsular hyperplasia nodules associated with benign prostatic hyperplasia
Chenghao Zheng, Yiping Zhu, Yifeng Jing, Shujie Xia
Current Urology.2025; 19(4): 247. CrossRef - The Surgical Treatment of Plantar Fibromatosis With Multiple Large Nodules on the Medial Aspect of the Left Sole With No Recurrence at the Five-Year Follow-Up
Christos Lyrtzis, Zoi Rafailia Sidiropoulou, Alexandra Arhonidu, Paissios Greige, Dimitrios Chytas, George Paraskevas
Cureus.2025;[Epub] CrossRef - A nodule on the foot
Michaela Thielen, Joshua Luciew, Brittany Strelow
JAAPA.2024; 37(4): 47. CrossRef - The Effects of Shock Wave Therapy on the Symptoms and Function of Individuals With Dupuytren Disease: A Systematic Review
Amid Yazdani, Parsa Nasri, Sadegh Baradaran Mahdavi
Archives of Physical Medicine and Rehabilitation.2024; 105(10): 1985. CrossRef - Palmar Fibromatosis, Updated Review With Relationship to the A1 Pulley, Trigger Finger, and Presence of the Sonographic Comb Sign
Cody A. Pepperday, Naveen Murthy, Sanjeev Kakar, Gavin A. McKenzie
Journal of Ultrasound in Medicine.2024; 43(12): 2281. CrossRef - Silent enemy of the hand: The Viking disease
Vera Stoiljković
Halo 194.2024; 30(3): 110. CrossRef - Prevalence of and Risk Factors for Adhesive Capsulitis of the Shoulder in Older Adults from Germany
Louis Jacob, Razak M. Gyasi, Ai Koyanagi, Josep Maria Haro, Lee Smith, Karel Kostev
Journal of Clinical Medicine.2023; 12(2): 669. CrossRef - Soft Tissue Masses of the Hand: A Review of Clinical Presentation and Imaging Features
Samuel AbuMoussa, Mona Pari Roshan, Felipe Ferreira Souza, Dane Daley, Andrew Rosenberg, Juan Pretell, Natalia Fullerton, Ty Subhawong
Current Oncology.2023; 30(2): 2032. CrossRef - Advanced Dupuytren Contracture
Jill Putnam
Hand Clinics.2023; 39(3): 455. CrossRef - Clinicopathological spectrum of lumps and bumps on the hand: A 5-year retrospective study
Kaumudi Konkay, Poola Neelima, Nugala Sindhura, Padmavathi Devi Chaganti
Journal of Clinical and Scientific Research.2023; 12(2): 81. CrossRef - Comorbid Medical Issues in X-Linked Ichthyosis
Lucija Brcic, Georgina H. Wren, Jack F.G. Underwood, George Kirov, William Davies
JID Innovations.2022; 2(3): 100109. CrossRef - The deep fascia and its role in chronic pain and pathological conditions: A review
Flemming Kondrup, Nathaly Gaudreault, Gabriel Venne
Clinical Anatomy.2022; 35(5): 649. CrossRef - Pacinian Corpuscles as a Diagnostic Clue of Ledderhose Disease—A Case Report and Mapping of Pacinian Corpuscles of the Sole
Jorge Feito, Ruth Esteban, María Lourdes García-Martínez, Francisco J. García-Alonso, Raquel Rodríguez-Martín, María Belén Rivas-Marcos, Juan L. Cobo, Benjamín Martín-Biedma, Manuel Lahoz, José A. Vega
Diagnostics.2022; 12(7): 1705. CrossRef
- APC-Mutated MUC4-Positive Fibroblastoma With Cytoplasmic β-Catenin: A Novel Variant Expanding Its Immunophenotypic Spectrum
- Clear Cell Adenocarcinoma Arising from Adenofibroma in a Patient with Endometriosis of the Ovary
- Inju Cho, Sung-Chul Lim
- J Pathol Transl Med. 2016;50(2):155-159. Published online October 26, 2015
- DOI: https://doi.org/10.4132/jptm.2015.08.07
- 12,563 View
- 137 Download
- 3 Web of Science
- 3 Crossref
-
Abstract
PDF
- Ovarian clear cell adenocarcinomas (CCACs) are frequently associated with endometriosis and, less often with clear cell adenofibromas (CCAFs). We encountered a case of ovarian CCAC arising from benign and borderline adenofibromas of the clear cell and endometrioid types with endometriosis in a 53-year-old woman. Regions of the adenofibromas showed transformation to CCAC and regions of the endometriosis showed atypical endometriotic cysts. This case demonstrates that CCAC can arise from CCAF or endometriosis.
-
Citations
Citations to this article as recorded by- Pure non-gestational ovarian choriocarcinoma in a postmenopausal woman coexisting with a clear cell adenofibroma and endometriosis foci: A case report and review of the literature
Ainhoa Ordoñez Arrillaga, Miguel Ángel Resano Abarzuza, Marta Rezola Bajineta, Begoña Aguiar Losada, Yessica P. Rodríguez-Velandia, Manuel Moreno Valladares, Iraide Bernal Simón, Ibon Jaunarena Marín, Irune Ruiz Díaz
Revista Española de Patología.2026; 59(1): 100856. CrossRef - Ovarian Clear Cell Adenofibroma of Low Malignant Potential Developing Into Clear Cell Adenocarcinoma
Zhiwei Yin, Stephen Peters, Ravi Chokshi, Debra Heller
International Journal of Surgical Pathology.2018; 26(6): 578. CrossRef - Origins based clinical and molecular complexities of epithelial ovarian cancer
Thingreila Muinao, Mintu Pal, Hari Prasanna Deka Boruah
International Journal of Biological Macromolecules.2018; 118: 1326. CrossRef
- Pure non-gestational ovarian choriocarcinoma in a postmenopausal woman coexisting with a clear cell adenofibroma and endometriosis foci: A case report and review of the literature
- WHO Grade IV Gliofibroma: A Grading Label Denoting Malignancy for an Otherwise Commonly Misinterpreted Neoplasm
- Paola A. Escalante Abril, Miguel Fdo. Salazar, Nubia L. López García, Mónica N. Madrazo Moya, Yadir U. Zamora Guerra, Yadira Gandhi Mata Mendoza, Erick Gómez Apo, Laura G. Chávez Macías
- J Pathol Transl Med. 2015;49(4):325-330. Published online June 17, 2015
- DOI: https://doi.org/10.4132/jptm.2015.05.20
- Correction in: J Pathol Transl Med 2015;49(6):538
- 11,369 View
- 78 Download
- 3 Web of Science
- 1 Crossref
-
Abstract
PDF
- We report a 50-year-old woman with no relevant clinical history who presented with headache and loss of memory. Magnetic resonance imaging showed a left parieto-temporal mass with annular enhancement after contrast media administration, rendering a radiological diagnosis of high-grade astrocytic neoplasm. Tumour sampling was performed but the patient ultimately died as a result of disease. Microscopically, the lesion had areas of glioblastoma mixed with a benign mesenchymal constituent; the former showed hypercellularity, endothelial proliferation, high mitotic activity and necrosis, while the latter showed fascicles of long spindle cells surrounded by collagen and reticulin fibers. With approximately 40 previously reported cases, gliofibroma is a rare neoplasm defined as either glio-desmoplastic or glial/benign mesenchymal. As shown in our case, its prognosis is apparently determined by the degree of anaplasia of the glial component.
-
Citations
Citations to this article as recorded by- Rare Pediatric Invasive Gliofibroma Has BRAFV600E Mutation and Transiently Responds to Targeted Therapy Before Progressive Clonal Evolution
Kristiyana Kaneva, Kee Kiat Yeo, Debra Hawes, Jianling Ji, Jaclyn A. Biegel, Marvin D. Nelson, Stefan Bluml, Mark D. Krieger, Anat Erdreich-Epstein
JCO Precision Oncology.2019; (3): 1. CrossRef
- Rare Pediatric Invasive Gliofibroma Has BRAFV600E Mutation and Transiently Responds to Targeted Therapy Before Progressive Clonal Evolution
- Abdominal Fibromatosis in a Young Child: A Case Study and Review of the Literature
- Hyun Hee Chu, Pyoung Han Hwang, Yeon Jun Jeong, Myoung Ja Chung
- Korean J Pathol. 2013;47(5):472-476. Published online October 25, 2013
- DOI: https://doi.org/10.4132/KoreanJPathol.2013.47.5.472
- 10,102 View
- 38 Download
- 3 Crossref
-
Abstract
PDF
Fibromatoses comprise many different entities of well-differentiated fibroblastic proliferation with variable collagen production and form a firm nodular mass. Abdominal fibromatosis is distinguishable from other forms of fibromatosis because of its location and its tendency to occur in women of childbearing age during or following pregnancy. Abdominal fibromatosis in children is an extremely rare condition. A 15-month-old boy presented with an abdominal wall mass that had recently increased in size. Mass excision was perfomed. The tumor was 4.3×4.1 cm and partly circumscribed. Histologically, the tumor was composed of parallel long fascicles of spindle-cells with a uniform appearance. The edges of the resected mass were infiltrative, and the surgical margins were positive. Mitotic figures were <1/10 high power fields. No cellular atypia or necrosis was present. The tumor cells were positive for vimentin and nuclear β-catenin staining.
-
Citations
Citations to this article as recorded by- A rare tumor of the large bowel in a young boy
Shyam Srinivasan, Soumitra Saha
Cancer Research, Statistics, and Treatment.2021; 4(4): 752. CrossRef - Uncommon abdominal wall mass in a young boy: Desmoid tumor
Levent Cankorkmaz, Mehmet H. Atalar, H. Reyhan Eğilmez
Cumhuriyet Medical Journal.2018; : 811. CrossRef - Lesiones ocupantes de espacio en pared abdominal (no herniaria). La visión del patólogo
Isidro Machado, Julia Cruz, Javier Lavernia, Fernando Carbonell
Revista Hispanoamericana de Hernia.2015; 3(3): 85. CrossRef
- A rare tumor of the large bowel in a young boy
- Papillary Carcinoma of the Thyroid Gland with Nodular Fasciitis-like Stroma
- Ki Yong Na, Hyun-Soo Kim, Ji-Youn Sung, Won Seo Park, Youn Wha Kim
- Korean J Pathol. 2013;47(2):167-171. Published online April 24, 2013
- DOI: https://doi.org/10.4132/KoreanJPathol.2013.47.2.167
- 11,368 View
- 54 Download
- 14 Crossref
-
Abstract
PDF
Papillary thyroid carcinoma with nodular fasciitis-like stroma (PTC-NFS) is a rare variant of PTC. The term 'PTC with fibromatosis-like stroma' has been used as a synonym to describe this variant. It is characterized by extensive proliferation of fibroblasts and myofibroblasts in the tumor stroma, which occurs in up to 80% of the tumors. We herein describe a case of PTC-NFS which developed in a 49-year-old woman with the demonstration of findings of ultrasonography, fine needle aspiration cytology and histological examination of the lesion. To characterize the stromal components, we investigated the expression of several immunohistochemical markers which have been shown to be expressed differently in nodular fasciitis (NF) and fibromatosis (FM). The immunostaining results demonstrated nuclear and cytoplasmic accumulation of β-catenin, cytoplasmic transforming growth factor-β expression and nuclear Smad expression in the stromal cells, suggesting that the stromal cells in this case have similar molecular profiles to those of FM rather than NF.
-
Citations
Citations to this article as recorded by- The Association of Interstitial Fibrosis and Ki67 Marker Expression in Papillary Thyroid Carcinoma with Histopathological Findings and Prognosis of Total Thyroidectomy
Osman Anıl Savaş, Selçuk Mercan, Amir Mahdi Akbari, Mehrdad Sheikhvatan
Journal of Clinical Medicine.2026; 15(14): 5703. CrossRef - Papillary thyroid carcinoma with nodular fasciitis-like stroma: An intriguing encounter
Pranjal Kalita, Biswajit Dey, Vandana Raphael, Gauranga Handique, Sungjemla Longkumer, Nikhil Choudhary
Thyroid Research and Practice.2025; 21(1): 41. CrossRef - Comparative Evaluation of Machine Learning Algorithms with Data Balancing Approach and Hyperparameter Tuning in Predicting Thyroid Disorder Recurrence
Darnell Ignasius, Rhyan David Levandra, Ramadhan Rakhmat Sani, Ika Novita Dewi
Jurnal Masyarakat Informatika.2025; 16(2): 284. CrossRef - Papillary thyroid carcinoma with desmoid-type fibromatosis: the clinicopathological features with characteristic imaging and molecular correlation requiring comprehensive treatment
Haining Huang, Lei Li, Xiaolong Liu, Lihua Zhao, Zhihong Cui, Renya Zhang, Shuai Chen
Human Pathology.2023; 136: 84. CrossRef - Papillary Thyroid Carcinoma with Desmoid-Type Fibromatosis: Review of Published Cases
Abdallah Roukain, Stefano La Rosa, Massimo Bongiovanni, Marie Nicod Lalonde, Valérie Cristina, Michael Montemurro, Stephane Cochet, Alexandra Luquain, Peter A. Kopp, Gerasimos P. Sykiotis
Cancers.2021; 13(17): 4482. CrossRef - Case of medullary thyroid carcinoma with desmoid‐type fibromatosis
Yoon Ah Cho, Young Lyun Oh
Pathology International.2020; 70(6): 364. CrossRef - SOX11 expression in a case of papillary thyroid carcinoma with fibromatosis/fasciitis-like stroma containing BRAF c.1799_1801delTGA and CTNNB1 c.133T>C mutations
Soon Boon Justin Wong, Min En Nga, Michal Michal, Tomas Vanecek, Ju Ee Seet, Fredrik Petersson
Virchows Archiv.2019; 475(4): 519. CrossRef - Papillary thyroid cancer with extrathyroidal extension of desmoid-type fibromatosis. A case report of an aggressive presentation of an uncommon pathologic entity
Eve M. Roth, Courtney E. Barrows, Michiya Nishino, Barry Sacks, Per-Olof Hasselgren, Benjamin C. James
International Journal of Surgery Case Reports.2019; 63(C): 5. CrossRef - Papillary thyroid carcinoma with nodular fasciitis-like stroma and β-catenin mutations should be renamed papillary thyroid carcinoma with desmoid-type fibromatosis
Caterina Rebecchini, Antoine Nobile, Simonetta Piana, Rossella Sarro, Bettina Bisig, Sykiotis P Gerasimos, Chiara Saglietti, Maurice Matter, Laura Marino, Massimo Bongiovanni
Modern Pathology.2017; 30(2): 236. CrossRef - Papillary thyroid carcinoma with desmoid-type fibromatosis: A clinical, pathological, and immunohistochemical study of 14 cases
Nami Takada, Mitsuyoshi Hirokawa, Masahiro Ito, Aki Ito, Ayana Suzuki, Miyoko Higuchi, Seiji Kuma, Toshitetsu Hayashi, Masao Kishikawa, Shuichi Horikawa, Akira Miyauchi
Endocrine Journal.2017; 64(10): 1017. CrossRef - A Case of Papillary Thyroid Carcinoma with Fasciitis-like Stroma
Toshihiko WAKU, Hiroshi SONOBE
Nihon Rinsho Geka Gakkai Zasshi (Journal of Japan Surgical Association).2016; 77(12): 2892. CrossRef - Stromal Modulation and its Role in the Diagnosis of Papillary Patterned Thyroid Lesions
Sahar Aly Daoud, Reham Shehab El Nemr Esmail, Amal Ahmed Hareedy, Abdullah Khalil
Asian Pacific Journal of Cancer Prevention.2015; 16(8): 3307. CrossRef - Papillary Thyroid Carcinoma With Nodular Fasciitis–Like Stroma
Paula S. Ginter, Theresa Scognamiglio
International Journal of Surgical Pathology.2015; 23(4): 305. CrossRef - Notch and TGF-β/Smad3 pathways are involved in the interaction between cancer cells and cancer-associated fibroblasts in papillary thyroid carcinoma
Jie Zhang, Yuan Wang, Dan Li, Shanghua Jing
Tumor Biology.2014; 35(1): 379. CrossRef
- The Association of Interstitial Fibrosis and Ki67 Marker Expression in Papillary Thyroid Carcinoma with Histopathological Findings and Prognosis of Total Thyroidectomy
- Intracranial Fibromatosis: A Case Report.
- Jeong Ju Lee, Jeoung Hun Kim, Shin Kwang Khang, Kyung Ja Cho, Jihun Kim
- Korean J Pathol. 2011;45:S89-S92.
- DOI: https://doi.org/10.4132/KoreanJPathol.2011.45.S1.S89
- 4,809 View
- 30 Download
- 1 Crossref
-
Abstract
PDF
- Fibromatosis can occur at various sites, but intracranial fibromatosis is exceptionally rare. Here, we report a case of intracranial fibromatosis arising in the suprasellar area of a 52-year-old woman who had undergone a surgery at that site. A computed tomography scan revealed a heavily calcified, highly enhancing, poorly demarcated mass in the left sellar area that extended into the left suprasellar, parasellar areas, and orbital apex and completely encased the left distal inferior cerebral artery. Histologic and immunohistochemical features were compatible with those of fibromatosis, although the cellularity was focally higher than usual. The etiology of extra-abdominal fibromatosis is unknown, but physical injuries such as trauma and irradiation have been reported to be associated with its occurrence. Although fibromatosis is rare in the intracranial area, it should be considered as a differential diagnosis when an intracranial mass occurs at a previously injured site.
-
Citations
Citations to this article as recorded by- Infantile Intracranial Aggressive Fibromatosis: Report of Two Cases with a Review of the Literature
Baocheng Wang, Jie Ma, Huiming Jin
Pediatric Neurosurgery.2012; 48(3): 181. CrossRef
- Infantile Intracranial Aggressive Fibromatosis: Report of Two Cases with a Review of the Literature
- A Fibroma with Cystic Change Developing in an Accessory Ovary: A Brief Case Report.
- Ae Ri Kim, Woo Jung Sung, Mi Jin Kim
- Korean J Pathol. 2011;45(3):319-321.
- DOI: https://doi.org/10.4132/KoreanJPathol.2011.45.3.319
- 4,655 View
- 37 Download
- 1 Crossref
-
Abstract
PDF
- Accessory ovaries are rare entities defined as small portions of ovarian tissue situated near, and connected to, the normally placed ovary. Tumors arising in accessory ovaries are extremely rare. In particular, a fibroma arising from an accessory ovary has not been reported in the literature. We describe such a case with a fibroma developing in an accessory ovary. A 53-year-old multiparous woman presented with abdominal pain for 2 months. Pelvic computed tomography revealed 11.0x8.0x6.0 cm sized cystic mass with a thick septal wall in right adnexa. The preoperative diagnosis was a borderline ovarian tumor. Furthermore, a laparotomy showed a cystic mass connected to the right ovary by stalk. The bilateral eutopic ovaries were completely normal. Histologically, an accessory ovary was replaced by a fibroma accompanied by extensive cystic change.
-
Citations
Citations to this article as recorded by- A Rare Case of Extensive Degeneration in Bilateral Ovarian Fibroma Mimicking Large Ovarian Cystadenoma
Tjokorda GA Pemayun, I Nyoman G Budiana
Journal of SAFOMS.2018; 6(2): 139. CrossRef
- A Rare Case of Extensive Degeneration in Bilateral Ovarian Fibroma Mimicking Large Ovarian Cystadenoma
- Use of Calretinin, CD56, and CD34 for Differential Diagnosis of Schwannoma and Neurofibroma.
- Ji Young Park, Hoon Park, Nam Jo Park, June Sik Park, Hyun Jung Sung, Sang Sook Lee
- Korean J Pathol. 2011;45(1):30-35.
- DOI: https://doi.org/10.4132/KoreanJPathol.2011.45.1.30
- 7,122 View
- 195 Download
- 23 Crossref
-
Abstract
PDF
- BACKGROUND
It is important to differentiate between schwannomas and neurofibromas for the cases in which the histopathologic features overlap. Depending on the tumor type, surgeons can decide on a treatment method and whether to preserve or sacrifice the nerve; the possibility of malignant transformation in the case of neurofibromas also needs to be considered.
METHODS
We studied 101 cases of schwannoma and 103 cases of neurofibroma. All the hematoxylin and eosin slides for these cases were reviewed, and tissue microarrays were prepared from the representative areas. Immunohistochemical analysis was performed using antibodies for S-100 protein, calretinin, CD56 and CD34.
RESULTS
All the tumors except 3 neurofibromas were positive for the S-100 protein. Calretinin was found in 26.7% of the schwannomas (27/101), but it was not found in any of the neurofibromas. CD56 was positive in 77.2% of the schwannomas (78/101) and in 9.8% of the neurofibromas (10/102). CD34 was positive in 42.5% of the schwannomas (43/101) and in 80.2% of the neurofibromas (81/101). Statistically, calretinin was significantly specific for schwannomas (p<0.001) and CD56 was also sensitive for these tumors (p<0.001). On the other hand, a CD34 expression seemed highly sensitive (p<0.001) for neurofibromas.
CONCLUSIONS
We concluded that combined immunohistochemical analysis for calretinin, CD56, and CD34 may be very useful for differentiating schwannomas from neurofibromas. -
Citations
Citations to this article as recorded by- Histomorphological Spectrum and Diagnostic Utility of S100, SOX10, CD34, Calretinin, and Ki67 in the Evaluation of Schwannomas: A Retrospective Study of 26 Cases in a Tertiary Health Institute, Zaria-Nigeria
Zainab Ali Adamu, Mikhail O. Buhari, Abdullahi Mohammed
Journal of West African College of Surgeons.2026; 16(2): 119. CrossRef - Ancient Schwannoma in the Nasal Cavity—A Rare Case Report With Brief Literature Review
Shikhar Chohan, Annmy Jose, Sufian Zaheer
Eye & ENT Research.2026;[Epub] CrossRef - Microcystic Pseudoglandular Cutaneous Neurofibroma: The First Japanese Case of a Rare Neurofibroma Variant
Shinichi Nakazato, Yasuyuki Fujita, Takashi Anan
The Journal of Dermatology.2025;[Epub] CrossRef - A rare case of giant intercostal nerve schwannoma
Bandari A. Ahmed, Ashley I. Simpson
International Surgery Journal.2025; 12(7): 1142. CrossRef - HISTOMORPHOLOGICAL SPECTRUM AND IMMUNOHISTOCHEMICAL EXPRESSION OF S100 AND CD56 AMONG TUMORS OF PERIPHERAL NERVES-A CROSS-SECTIONAL STUDY
KARTHIK SIGAMANI, SHOBANA B, NAYANA CHANDRAN
Asian Journal of Pharmaceutical and Clinical Research.2025; : 142. CrossRef - The skin's secret script: fingerprints of CD34 and the dolphin dance of Schwann cells
Vijay Joshi, Ketki Bhoite, Vidya Kharkar, Rajiv Joshi, Surupa Das
International Journal of Research in Dermatology.2025; 11(6): 541. CrossRef - Breast schwannoma: review of entity and differential diagnosis
Sandra Ixchel Sanchez, Ashley Cimino-Mathews
Journal of Pathology and Translational Medicine.2025; 59(6): 353. CrossRef - Primary Pulmonary Neurofibroma: Diagnostic Approach to a Common Tumor at an Uncommon Site – A Case Report
Rakesh Kumar Gupta, Bhoomika Kaushik, Shaurya Vijayran, Ranganath Ganga, Mudalsha Ravina
SN Comprehensive Clinical Medicine.2025;[Epub] CrossRef - Ancient cervical vagal schwannoma and an interposition great auricular to vagus nerve graft
Nicholas Figaro, Rickhi Ramoutar, David Richards, Rodolfo Arozarena, Mala Geelal, Solaiman Juman
Edorium Journal of Otolaryngology.2024; 5(2): 1. CrossRef - Lung schwannomas, an unusual entity
Nazia M. Walvir, Rumana H. Makhdoomi, Meesa Zargar, Aiffa Aiman, Shadab Maqsood
Lung India.2023; 40(1): 70. CrossRef - A Spectrum of Histomorphological and Immunohistochemical Expression Profiles of S-100, CD56 and Calretinin in Benign Peripheral Nerve Sheath Tumours
Poornima Jaiswal, Anand CD, Jaison Jacob John
Cureus.2023;[Epub] CrossRef - An unusual diffuse CD34 staining in an olfactory groove cellular schwannoma: Case report
Marios Theologou, Jorge D. Perez Ruiz, Panagiotis Varoutis, Nicolaos Flaris, Nikolaos `Skoulios
Archivos de Neurociencias.2023;[Epub] CrossRef - An unusual diffuse CD34 staining in an olfactory groove cellular schwannoma: Case report
Marios Theologou, Jorge D. Perez Ruiz, Panagiotis Varoutis, Nicolaos Flaris, Nikolaos `Skoulios
Archivos de Neurociencias.2023;[Epub] CrossRef - Intranodal Neurofibroma: A Case Report and Literature Review
Steven H. Adams, Tara L. Huston, Daniel Lozeau
The American Journal of Dermatopathology.2022; 44(4): 306. CrossRef - A rare case of pseudoglandular schwannoma
Fadime Eda GÖKALP SATICI, Hamide SAYAR
Journal of Surgery and Medicine.2022; 6(4): 1. CrossRef - Gastric Schwannoma as an Important and Infrequent Differential Diagnosis of Gastric Mesenchymal Tumours: A Case Report and Review of Literature
Abdalla Saad Abdalla Al-Zawi, Salma Lahmadi, Saman Jalilzadeh Afshari, Ipshita Kak, Salem Alowami
Cureus.2022;[Epub] CrossRef - Spindle cell proliferations of the sigmoid colon, rectum and anus: a review with emphasis on perineurioma
Patrice Grech, John B Schofield
Histopathology.2020; 76(3): 342. CrossRef - Large retroperitoneal schwannoma: a rare cause of chronic back pain
Milan Radojkovic, Dragan Mihailovic, Miroslav Stojanovic, Danijela Radojković
Journal of International Medical Research.2018; 46(8): 3404. CrossRef - Nasal Septal Schwannoma
Hyun Jin Min, Seok Chan Hong, Kyung Soo Kim
Journal of Craniofacial Surgery.2017; 28(1): e97. CrossRef - Neurofibroma of the Colon: A Diagnostic Mimicker of Gastrointestinal Stromal Tumor
Soomin Ahn, Choon Sik Chung, Kyoung-Mee Kim
Case Reports in Gastroenterology.2016; 10(3): 674. CrossRef - Solitary Epibulbar Neurofibroma in Older Adult Patients
Thais Shiota Tanaka, Victor M. Elner, Hakan Demirci
Cornea.2015; 34(4): 475. CrossRef - Syncytial nuclear aggregates in normal placenta show increased nuclear condensation, but apoptosis and cytoskeletal redistribution are uncommon
S.J. Coleman, L. Gerza, C.J.P. Jones, C.P. Sibley, J.D. Aplin, A.E.P. Heazell
Placenta.2013; 34(5): 449. CrossRef - Analysis of syncytial nuclear aggregates in preeclampsia shows increased sectioning artefacts and decreased inter-villous bridges compared to healthy placentas
S.J. Calvert, C.J.P. Jones, C.P. Sibley, J.D. Aplin, A.E.P. Heazell
Placenta.2013; 34(12): 1251. CrossRef
- Histomorphological Spectrum and Diagnostic Utility of S100, SOX10, CD34, Calretinin, and Ki67 in the Evaluation of Schwannomas: A Retrospective Study of 26 Cases in a Tertiary Health Institute, Zaria-Nigeria
- Lipofibromatosis: A Case Report.
- Tae Eun Kim, Tae Jung Kim, Youn Soo Lee, Chang Suk Kang, Sang In Shim, Kyo Young Lee
- Korean J Pathol. 2011;45(1):106-110.
- DOI: https://doi.org/10.4132/KoreanJPathol.2011.45.1.106
- 3,958 View
- 59 Download
-
Abstract
PDF
- Lipofibromatosis is a recently described rare benign fibrofatty tumor of childhood. It typically forms as an ill defined, slowly growing, painless mass. We present here the case of lipofibromatosis that occurred in a 21-year-old male who had complained of a bulging enlarged mass involving the right thigh and prepatella area for the previous 1 year. Magnetic resonance imaging showed an ill-defined reticular infiltration in the subcutaneous layer with subtle linear enhancement and high T2 signal intensity. The mass was surgically excised and it displayed an 11.0x5.5x1.5 cm-sized adipose appearance without encapsulation. Microscopically, the tumor was composed of alternating streaks of mature adipose tissue and a fibroblastic component that mainly involved the septa of adipose tissue. On immunohistochemical study, the fibroblastic component was positive for S-100, CD99, CD34, actin and bcl-2. He has shown an eventful recovery for 6 months after surgery.
- Chondromyxoid Fibroma of the Ethmoid Sinus Complicated by a Brain Abscess: A Case Report and Literature Review.
- Kyu Yeoun Won, Juhie Lee, Youn Wha Kim, Eui Jong Kim, Sung Wan Kim, Yong Koo Park
- Korean J Pathol. 2010;44(5):547-550.
- DOI: https://doi.org/10.4132/KoreanJPathol.2010.44.5.547
- 4,618 View
- 26 Download
- 2 Crossref
-
Abstract
PDF
- Chondromyxoid fibroma (CMF) is a relatively rare bone tumor that was first described by Jaffe and Lichtenstein in 1948. CMF of the sinonasal tract is very rare. A 28-year-old male presented with long-standing, intermittent, pulsatile pain in the right temporal area. A computed tomography scan showed a 20 x 19 mm round, bony density in the right ethmoid sinus with fluid collection in the ethmoid and frontal sinuses. Additionally, a cystic lesion with surrounding edema was found in the right frontal lobe. The patient underwent a partial ethmoidectomy and frontostomy. A histological examination showed polygonal and stellate cells in a myxoid and chondroid background with a pattern of lobulation and plaque-like calcification. The bone lesion was revealed as a CMF of the ethmoidal sinus, and the frontal lobe cystic lesion was a brain abscess associated with the CMF. We present the case of a CMF of the ethmoid sinus complicated by a brain abscess.
-
Citations
Citations to this article as recorded by- Juxtacortical chondromyxoid fibroma in the small bones: two cases with unusual location and a literature review
Sun-Ju Oh, So Hak Chung
Journal of Pathology and Translational Medicine.2022; 56(3): 157. CrossRef - Treatment of cryotherapy and orthotopic transplantation following chondromyxoid fibroma of zygomatic bone
Zhi-Chao Zhu, Yi-Fei Yang, Xu Yang, Yan Liu, Yi-nan Cheng, Zhao-Yao Sun, Tian-Shu Xu, Wen-Jun Yang
Medicine.2018; 97(31): e11707. CrossRef
- Juxtacortical chondromyxoid fibroma in the small bones: two cases with unusual location and a literature review
- Calcifying Aponeurotic Fibroma of the Elbow: A Case Report.
- Mee Hye Oh, Eun Ah Jung, Ji Hye Lee, Hyun Deuk Cho, Jong Kyu Han, Yong Koo Park
- Korean J Pathol. 2009;43(1):75-78.
- DOI: https://doi.org/10.4132/KoreanJPathol.2009.43.1.75
- 4,745 View
- 35 Download
-
Abstract
PDF
- Calcifying aponeurotic fibroma is a rare soft tissue tumor that mostly occurs in the distal extremities of children and adolescents. We report here on a case of calcifying aponeurotic fibroma of the right elbow in an 8-year-old boy, and the tumor was diagnosed by surgical excision. The patient complained of painless swelling and mild limitation of the range of motion of the elbow joint. Radiologically, the mass was ill-defined and showed stippled calcification with shallow bony erosion. Microscopically, the tumor was composed of spindle cells with nodular deposits of hyalination and calcification, and these deposits were surrounded by palisading polygonal plump cells. Immunohistochemically, the tumor showed a diffuse positive expression for CD99 and negativity for smooth muscle actin, S-100 protein and CD34. The patient has been well with no signs of recurrence during the 42 months after surgery.
- Juvenile Cellular Adenofibroma of Breast: A case report.
- Je G Chi, Yeon Lim Suh
- Korean J Pathol. 1989;23(2):269-272.
- 2,245 View
- 16 Download
-
Abstract
PDF
- Juvenile cellular adenofibroma of the breast is a unique neoplasm of the breast that should be differentiated from other important benign and malignant lesions of the juvenile breasts. We report a case with it's characteristic clinical, gross and histological features. The tumor was in the right breast with the size of 20 cm in maximum extent. This patient was also associated with hemihypertrophy of the right side. Microscopically the masses were characterized by prominent stromal cellularity associated with pericanalicular duct proliferation.
- Elastofibroma: Report of a case.
- Hye Kyung Lee, Kwang Min Lee, Dong Kyu Chung, Eul Sam Chung
- Korean J Pathol. 1992;26(6):635-637.
- 2,005 View
- 12 Download
-
Abstract
PDF
- Elastofibroma is a rare entity of slowly-growing, solid, ill-defined fibroblastic mass occurring almost exclusively in elderly persons and arising mainly from the connective tissue between the lower portion of the scapula and the chest wall. This entity has been considered to be a degenerative pseudo-neoplastic process caused by mechanical friction. We report an additional case of elastofibroma removed from the subscapular region of a 58-year-old woman. Microscopically the tumor was made up of a mixture of swollen eosinophilic collagen and elastic fibers occasionally associated with fibroblasts and mature fat cells. The elastic fibers showed a degenerated beaded appearance or were fragmented into small globules or droplets in a linear pattern.
- Renomedullary Interstitial Cell Tumor.
- Eon Sub Park, Mi Kyung Kim, Jae Hyung Yoo, Kye Yong Song
- Korean J Pathol. 1989;23(3):371-373.
- 2,557 View
- 25 Download
-
Abstract
PDF
- We present an ultrastructure of an incidentally found renomedullary interstitial tumor also called as medullary fibroma in a 77 year-old female who had a metastatic adenocarcinoma of colon to the ureter. This tumor was a small and grayish white nodule in renal medulla, measuring 0.4 x 0.4 cm. Microscopically the tumor composed of spindle cells, with some vacuolation and intercellular collagen fibers. The electron microscopic observation of the spindle cells reveal that nuclei are spindle to oval shape and cytoplasm contain abundant rough endoplasmic reticulum, polyribosome without microfilaments and cisterna like structures supporting that the renomedullary interstitial cell tumor is renal interstitial cell origin than fibroblasts.
- Development of Desmoid and Mesenteric Fibromas following Total Colectomy for Adenomatous Polyposis Coli in Gardner's syndrome.
- Jung Hee Cho, Yong Il Kim
- Korean J Pathol. 1989;23(4):465-469.
- 1,959 View
- 11 Download
-
Abstract
PDF
- We describe a case of polyposis coli, which was followed by development of desmoid in the rectus adbominis muscle and fibromas in the mesentery during an interval of two years. This case supports the hypothesis that, in Garder's syndrome, the traumatic injury by colectomy triggers an unusual fibrous proliferation in the peritoneal cavity and incision site under the possible genetic basis.
- Juvenile Ossifying Fibroma: A Clinicopathologic Study of 8 Cases and Comparison with Craniofacial Fibro-osseous Lesions.
- Sohyung Park, Bong Jae Lee, Jeong Hyun Lee, Kyung Ja Cho
- Korean J Pathol. 2007;41(6):373-379.
- 2,254 View
- 28 Download
-
Abstract
PDF
- BACKGROUND
Juvenile ossifying fibroma (JOF) is defined as a variant of the ossifying fibroma, and the latter includes juvenile trabecular ossifying fibroma (JTOF) and juvenile psammomatoid ossifying fibroma (JPOF). JOF can be distinguished from other craniofacial fibroosseous lesions by its tendency to recur and its clinical mimicry of malignant bone tumors, but some clinical and histological features of JOF overlap with the other fibro-osseous lesions as well. We aimed to identify the clinicopathologic definition of JOF.
METHODS
Forty-two cases of fibro-osseous lesions were reviewed and they were classified into JOF, fibrous dysplasia (FD) and ossifying fibroma (OF).
RESULTS
JTOF had long, slender and anastomosing trabeculae of osteoid in a fibrocellular stroma, and JPOF had small ossicles resembling psammoma bodies with a thick collagenous rim in the fibrous stroma, which are features that differ from those of FD and OF. Radiologically, JOF and OF showed a well-defined lesion but FD exhibited an ill-defined lesion. Clinically, the average age of the JOF patients was the youngest, followed by OF and FD. For JOF, three cases had rapid growth and two others showed recurrences. JOF mainly occurred in the paranasal sinuses, OF in the mandible and FD in any craniofacial bone.
CONCLUSION
We demonstrated the distinct characteristics of JOF and these features may be helpful for the diagnosis and management of this malady.
- Cellular Angiofibroma of the Vulva: A Report of Three Cases.
- Hye Jeong Choi, Sung Nam Kim, Kyu Rae Kim
- Korean J Pathol. 2001;35(3):259-262.
- 2,381 View
- 29 Download
-
Abstract
PDF
- Cellular angiofibroma is a recently described, distinctive soft tissue tumor of the vulvovaginal region which is characterized by small, well-circumscribed tumors with fibroblastic differentiation. We report three cases of cellular angiofibroma of the vulva in middle-aged women. All three patients presented with painless swelling in the labium majora. The age of the three patients ranged from 43 to 56 years old (mean: 48 years old) and the size of the tumor ranged from 2 to 5 cm. The microscopic appearance was characterized by a cellular, well-circumscribed mass composed of uniform, bland, spindle stromal cells, numerous thick-walled, hyalinized vessels, and a scarce component of mature adipocytes. Immunohistochemical stains of the tumor cells show positivity for vimentin but negativity for smooth muscle actin, S-100 protein, desmin, factor VIII-related antigen and epithelial membrane antigen. The tumor should be differentiated from aggressive angiomyxoma and angiomyofibroblastoma because of its different clinicopathologic features, cells of origin and immunohistochemical findings.
- A Case of Solitary Cutaneous Myofibroma of the Thigh in An Adult.
- Jung Hwan Park, Chang Woo Lee, Young Chae Chu, Moon Hyang Park
- Korean J Pathol. 2001;35(4):354-356.
- 2,407 View
- 33 Download
-
Abstract
PDF
- Adult solitary cutaneous myofibroma is a recently described benign neoplasm of the skin or subcutis, representing the adult counterpart of infantile myofibroblastoma. The histologic and immunohistochemical features of a 21-year-old woman with a solitary brownish, mildly tender nodule on her right thigh are reported here. The nodule had been present for a duration of 3 years. It showed a nodular dermal mass with an irregular margin. The lesion consisted of interlacing bundles of spindle cells which were positive for smooth muscle actin, muscle specific actin and vimentin. Immunohistochemical stainings for desmin, S-100 protein, CD 34 and CD 68 were negative. Cutaneous myofibroma in an adult is a distinct entity of benign neoplasm.
- Bilateral Elastofibroma: Report of a case.
- Sung Chul Lim, Mi Sook Lee, You Kyung Jeong, Yun Shin Kim, Hyun Jong Park, Mi Ja Lee
- Korean J Pathol. 1997;31(6):589-591.
- 2,251 View
- 14 Download
-
Abstract
PDF
- Elastofibroma is a rare benign tumor-like condition manifesting as firm and spherical mass with poorly circumscribed margins of fibroelastic tissue, occuring in the subscapular region or the chest wall of elderly persons. It is not a true neoplasm but rather a reactive or degenerative process causing abnormal elastogenesis. It is unilateral in the majority of cases and the right side is affected more commonly than the left. We report a case of bilateral elastofibromas removed from both subscapular regions of a 73-year-old female farmer. She was presented with tender masses on the bilateral subscapular areas for seven years. Microscopically, it consisted of a mixture of intertwining broad eosinophilic collagen bundles and elastic fibers associated with a few fibroblasts and mature fat cells. The elastic fibers had a degenerated beaded appearance or were fragmented into serrated globules in a linear arrangement.
- Infantile Myofibromatosis(Congenital Generalized Fibromatosis): Associated with multiple congenital malformations and basaloid follicular hamartomas in the skin.
- Eun Sook Nam, Yoo Hun Kim, Han Kyeom Kim, Insun Kim, Je Geun Chi
- Korean J Pathol. 1995;29(6):776-782.
- 2,447 View
- 19 Download
-
Abstract
PDF
- Infantile myofibromatosis with systemic involvement is a very rare disease and is characterized by numerous nodules composed of spindle cells of a myofibroblastic nature. There are often disseminated throughout the subcutis, muscle, skeleton and viscera. We report an autopsy case of infantile myofibromatosis in a stillborn female fetus of 32 weeks of gestation. The nodules, Imm to 2 cm, were found over the whole body and viscera. The involved viscera were the heart, tongue, esophagus, gastrointestinal tract, portal areas of the liver, spleen anc pancreas. There were also associated malformations, viz., frontal meningoencephalocele, flexion defer-mities, syndactyly, cleft palate, agenesis of corpus callosum, pachygyria, diaphragmatic hemia, renal hypoplasia, etc. Multiple basaloid follicular hamartomas of the skin were noted on the face and extremeties. There are no previous reports in the literature of infantile myofibromatosis in conjunction with the above skin lesion and congenital malformations.
- Dendritic Myxofibrolipoma.
- Sung Nam Kim, Kye Hyun Kwon, Yeon Lim Suh
- Korean J Pathol. 2001;35(5):447-450.
- 2,431 View
- 24 Download
-
Abstract
PDF
- Dendritic myxofibrolipoma is a recently described disease entity that represents a distinctive benign soft tissue neoplasm showing the combined features of spindle cell lipoma and the solitary fibrous tumor. Immunohistochemical stains reveal a strong positivity for vimentin, CD34 and bcl-2, which highlight the dendritic nature of the tumor cells by demonstrating slender complex cytoplasmic prolongations. There have been 12 cases of dendritic myxofibrolipomas reported in literature. In Korea, none of the cases have been described. We report such a case with a 28-year-old man who had a palpable subcutaneous mass on his right shoulder for 4 months. Grossly, the removed mass measured 11X7X5 cm and appeared to be a well-encapsulated, lipomatous tumor with marked myxoid appearance. Microscopically, this tumor consisted of spindle cells admixed with dense collagen fibers and mature adipocytes in abundant myxoid stroma with high vascularity. Immunohistochemically, the tumor cells were strongly reactive for vimentin and CD34 and weakly reactive for bcl-2, and negative for S-100 protein.
- Optic Nerve Glioma with Neurofibromatosis.
- Na Hye Myong, Seung Sook Lee, Yun Lim Shu, Je G Chi
- Korean J Pathol. 1993;27(5):524-530.
- 2,386 View
- 33 Download
-
Abstract
PDF
- Optic nerve gliomas manifest either as a solitary form or a component of von Recklinghausen's neurofibromatosis. The reported incidence of coexistence with neurofibromatosis varies from 12% to 70%. Usually there are no significant cytological differences between the gliomas that accomapny the disease and those that are deemed to be solitary manifestations. The only possible difference between them is the apparently more common association, with the former, of extensive arachnoid hyperplasia and of a more florid local gliomatous infiltration into the leptomeninges, altogether resulting in perineural thickening. Our cases were 8 and 6 years old girls, respectively, presented with slowly progressive proptosis for 4 years and visual disturbance for 2.5 months. There were multiple cafe au lait spots on their trunks, and case 2 showed Lisch nodules in the iris. MRI of brain revealed unilateral optic nerve thickening with involvement of chiasm or multiple intracranial lesions. Resection of optic nerve tumor was performed. Microscopically, variable degree of tumorous change was seen. Most typically enlarged area was composed of intraneural and perineural portions surrounded by a layer of intact dura. Intraneural tumor revealed proliferation of elongated, spindle-shaped pilocytic astrocytes in intertrabecular spaces and distention of the pial septa with fibrovascular thickening. Another segment had areas with reactive gliosis. Perineural tissue was considerably thickened and, associated with proliferation of meningothelial cells and fibroblasts intermingled with astrocytes and Rosenthal fibers. There were increase of the optic nerve diameter and distention of the overlying dura. Foci of arachnoid cell hyperplasia were noted in both cases, although differed in degree. Immunohistochemically, the tumor cells expressed glial fibrillary acidic protein in intraneural and perineural portions particularly in case 2.
- Fibromatosis of the Breast: A Case Report.
- Hyun Joong Kim, Kyung Hwa Lee, Jo Heon Kim, Min Keun Shim, Ji Shin Lee, Chan Choi
- Korean J Pathol. 2005;39(2):137-139.
- 3,196 View
- 21 Download
-
Abstract
PDF
- Fibromatosis of the breast is a rare tumor. We describe here a case of mammary fibromatosis in a 37-year-old woman. The mass from the right breast was 3 cm at the greatest dimension. The lesion was poorly circumscribed, firm and white-gray on the cut surface. Histologically, the lesion infiltrated into the lobules of the breast, and the tumor was composed of relatively uniform fibroblasts and collagen. Neither mitotic activity nor cellular atypia was seen. On the immunohistochemistry, the cells were positive for vimentin and they were focally positive for smooth muscle actin. Staining results for estrogen receptor and progesterone receptor were negative.
- Malignant Endometrioid Adenofibroma of the Ovary: A case report.
- Tae Jung Jang, Soon Hee Jung, Kyu Rae Kim, Hoguen Kim
- Korean J Pathol. 1990;24(4):497-501.
- 3,053 View
- 70 Download
-
Abstract
PDF
- Ovarian endometrioid adenofibroma is rare and characterized by prominent stroma. Its histologic classification is controversial but the malignant counterpart is distinguished from the borderline by the presence of confluent growth pattern of epithelium with invasion of the stroma by the endometrioid cells. A fifty-year-old woman was admitted with one month history of abdominal enlargement. Total abdominal hysterectomy with bilateral salpingo-oophorectomy was performed under the clinical diagnosis of ovarian malignancy. Grossly, the righy ovary had round, encapsulated, solid and whitish gray mass which measured 9 cm in the greatest dimension and showed peripheral small cysts. Microscopic examination revealed that the tumor consisted of endometria type glands set in fibrous stroma. The glands varied from tubules to cysts and the lining cells showed complicated architectural pattern with occasional papillary infoldings, atypical mitosis and malignant nuclear characteristics. Some cysts of glands showed intraluminal mucin products. Stromal invasions by individual epithelial cells showing malignant characteristics were often found. A brief summary of the histopathologic aspect of this tumor is presented together with review of literatures.
- Cardiac Fibroma of the Ventricular Septum: A case report.
- Byung Tae Park, Se Jin Jang, Moon Hyang Park, Jung Dal Lee, Hyo Jin Lee
- Korean J Pathol. 1991;25(1):37-41.
- 2,249 View
- 20 Download
-
Abstract
PDF
- This is an autopsy case of a 6 month old girl who suddenly died of respiratory distress during sleep. She had suffered from mild but frequent episodes of common cold and was treated for eczema for several days. At autopsy, the heart was enlarged and weighed 100 gm. A firm and gray-white tumor, measuring 4.5 x 3.8 x 2.8 cm, was located in the interventricular septum and encroached upon the wall of left ventricle. The mass was well demarcated but was not encapsulated. Neither necrosis nor calcification was present. Microscopically the tumor was composed of haphazardly arranged bundles of collagen fibers and fibroblasts. Myocardial cells are intermingled with the fibroblasts at the margin of the tumor. Massive edema of the lung and congestion of the liver and spleen were pronounced.
- Fibromatosis of the Parotid Gland: A Case Report.
- Dae Su Kim, Chulhwan Kim, Insun Kim
- Korean J Pathol. 2002;36(2):112-114.
- 2,637 View
- 30 Download
-
Abstract
PDF
- A 51-year-old woman was presented with a palpable mass in the infraauricular area that had existed for 4-months. The mass from the superficial lobe of the parotid gland was 2.7 cm in the greatest dimension and was ill-defined with rubbery, homogeneous, and fibrotic appearance. Histologically, the lesion was infiltrative into the lobules of the paratid gland, and was composed of a proliferation of spindle or stellate cells with cellular and sclerotic areas. On immunohistochemistry, the cells were only positive for vimentin and focally for smooth muscle actin. Differential diagnosis from nerve sheath tumors and solitary fibrous tumors involving the parotid gland was emphasized.
- Trichogerminoma: A case report.
- Sung Suk Paeng, Jin Hee Sohn, Duck Hwan Kim, Hee Jin Chang, Jung Il Suh, Kye Yong Song
- Korean J Pathol. 1996;30(4):340-343.
- 2,243 View
- 30 Download
-
Abstract
PDF
- Though trichogenic tumors were classified as trichoblastoma, trichoblastic fibroma, trichogenic trichoblastoma and trichogenic myxoma by Headington(1970), their true classification depends upon the epithelial and mesodermal component as well as evidence of their induction. Because of the rarity of hair germ cell tumors their classification is still controversial. In this report, we describe a case of trichogerminoma which is not included in the above classification. The trichogerminoma was first described by Sau et al. in 1992 and characterized by its morphologic pattern of germinal centers and lymphoid follicle-like structures in the nests of trichoblasts. Herein we reporte a tumor which arose on the skin on the back of a 51-year-old man and presented as a sharply circumscribed mass(4.5x2.0x1.5 cm) involving both the dermal and subcutaneous tissues without any epidermal connection. The tumor had many germinal center-like structures in the basaloid trichoblasts. Lobular cell nests were separated by variable amounts of stroma, but no horn cyst were noted. The germinal center-like cells showed early differentiation of hairs, resembling early hair bulbs. Trichogerminoma is considered to be a type of tumor located between trichoblastoma and trichoblastic fibroma.
- A Case of Ocular Neurofibromatosis.
- Je G Chi, In Ae Park
- Korean J Pathol. 1987;21(1):62-65.
- 2,204 View
- 20 Download
-
Abstract
PDF
- Neurofibromatosis is one of the neurocristopathies that involve many system or tissues forming various types of lesion. Almost every tissue or organ can be involved by this disease. However, the eyeball itself is very rarely affected by this process. The findings seen in our case indicate the diversity of lesions in neurofibromatosis, and also suggest hamartomatous nature. Heterotopic ganglion cells and glial cells in uveal tract are not easily understood. We reported here a case of intra-occular neurofibromatosis with its characteristic involvement of the uveal tract, in a 21 year old female. Her ocular symptoms began at her age of 11 as poor vision and were slowly progressive together with multiple facial neurofibromas. The involved left eyeball showed many ganglioneuroglial cell nests in iris, ciliary body and retina. Minute plexiform neurofibromas were also seen in small nerve twigs around the eyeball.
- Fine Needle Aspiration Cytologic Findings of Fibromatosis Colli: A Report of Three Cases.
- In Suh Park, Lucia Kim, Suk Jin Choi, Jee Young Han, Joon Mee Kim, Young Chae Chu, Sun Geun Choi
- J Pathol Transl Med. 2005;16(1):61-65.
- 2,623 View
- 24 Download
-
Abstract
PDF
- Fibromatosis colli is a benign fibrous tissue proliferation of sternocleidomastoid muscle, which is usually observed during the first month of life, often associated with congenital torticollis. It should be differentiated from other neck masses in infants because the usual initial treatment of fibromatosis colli is conservative management and invasive therapy should be avoided. Fine needle aspiration cytology provides an excellent minimally invasive diagnostic way for evaluation of infantile neck masses. We describe three cases of fibromatosis colli diagnosed by fine needle aspiration cytology. All of them were younger than one month and presented as a neck mass. Clinical impressions were malignant tumors in two cases and fibromatosis colli in one case. Fine needle aspiration cytology revealed benign and mature fibroblasts and atrophic striated muscle fibers. The cytologic diagnosis was fibromatosis colli or benign fibous lesion.
- Cardiac Fibroma: A surgically excised case.
- Ho Jung Lee, Gyung Ub Gong, Jay Won Lee, Jae Gon Go, In Chul Lee
- Korean J Pathol. 1996;30(6):544-547.
- 2,201 View
- 12 Download
-
Abstract
PDF
- Primary cardiac tumors in infancy and childhood are rare, with fibromas being the second most common tumor after rhabdomyomas. Although cardiac fibromas are characteristically benign intramural tumors, they may exhibit exhibit expansile growth resulting in obstruction, valvular dysfunction, as well as other problems so early diagnosis and successful surgical excision are important. We report a case of cardiac fibroma in a 2 month-old male infant. He presented with generalized cyanosis from birth. Echocardiography showed oval round large mass filing the right atrium and ventricle which infiltrated into the lateral wall of the ventricle. Partial excision of the tumor was done after another echocardiogram showed a pericardial effusion and restriction of blood flow to the right ventricle due to the tumor. The resected tumor was ovoid, gray-tan, slightly firm and measuring 5x3x2.5 cm. Histologically, the tumor was composed of spindle-shaped fibroblasts and hyalinized fibrous tissue interdigitating with the surrounding myocardium.
- Benign Lymphoepithelial Cyst: A case report.
- Jin Haeng Chung, Gyeong Hoon Kang, Je G Chi
- Korean J Pathol. 1996;30(6):551-553.
- 2,712 View
- 20 Download
-
Abstract
PDF
- An intraparotid benign lymphoepithelial cyst is a rare disease characterized by unilateral painless swelling of parotid region. The histogenesis is controversial. Surgical excision is recommended for diagnosis and curative treatment. We present a case of benign lymphoepithelial cyst arising in a patient with neurofibromatosis. A 46-year-old woman presented with a slowly growing multilocular cystic mass in the left cheek. The cystic mass measured 4 cm in maximal outer diameter and the cystic wall was thick and yellowish pale to gray, soft with well circumscribed margin. Microscopically, the multilocular cyst was lined by stratified squamous epithelium for the most part and underlying lymphoid tissue aggregates with follicles and sharply demarcated from adjacent salivary parenchyma which is of normal appearance and without lymphoid aggregates. Since this lesion is absolutely benign, it is important to separate this benign cyst from cystic salivary gland tumors.
- Gastrointestinal Stromal Tumors associated with Neurofibromatosis Type I: A Report of Two Cases.
- Joo Heon Kim, Ock Seong In, Seong Kyu Lee, Haing Woon Baik, Seong Ho Kim, Dong Wook Kang, Kyung Hee Kim, Mee Ja Park, Yong Il Kim
- Korean J Pathol. 2006;40(2):137-141.
- 2,449 View
- 23 Download
-
Abstract
PDF
- Gastrointestinal stromal tumor (GIST) is the most common non-epithelial neoplasm arising in the gastrointestinal tract, but this tumor is rarely seen in association with type 1 neurofibromatosis (NF-1). We report here on two cases of multiple GISTs of the small intestine that occurred in NF-1 patients. We also analyzed the mutations of c-kit exons 9, 11, 13 and 17 and the plateletderived growth factor receptor-alpha (PDGFRA) exons 12 and 18 in two GIST patients. Histologically, the NF-1-associated GISTs were similar to those of non-the NF-1 GISTs, but they characteristically revealed hyperplastic interstitial cells of Cajal around the GISTs. Immunohistochemically, these tumors showed strong co-expressions of CD117 and CD34. The molecular genetic analysis of the GISTs showed that all of the c-kit and PDGFRA exons that were analyzed in the GISTs of the two patients were the wild-type, suggesting a limited role for the c-kit and PDGFRA mutations in the tumorigenesis of NF-1-associated GISTs.
- Juvenile Hyaline Fibromatosis in an Adult.
- Young A Kim, Seoung Wan Chae, Chong Jai Kim, Je G Chi
- Korean J Pathol. 2000;34(3):239-242.
- 2,675 View
- 42 Download
-
Abstract
PDF
- Juvenile hyaline fibromatosis is a rare disorder probably inherited as an autosomal recessive trait. It is characterized by multiple slowly growing subcutaneous nodules, hypertrophy of gingiva, flexion contracture, and radiolucent bone destruction. The histological features of the tumor-like lesions are characterized by the deposition of amorphous hyaline material in which spindle shaped cells are embedded. We report a case of juvenile hyaline fibromatosis in a 26 year-old-woman. She had multiple subcutaneous nodules in scalp, ear, forearms, right knee, and back. Surgical excision of the tumors in the scalp and ear was done. The largest one measured 13 9 6 cm, and had homogeneous, grayish yellow cut surface with calcification. Light microscopic examination showed abundant eosinophilic hyaline material with extensive calcification and metaplastic bone formation. Spindle cells were rarely observed at the periphery of the tumor. Hyaline matrix was PAS positive, diastase resistant, and alcian blue negative. Scattered spindle cells were positive for vimentin but negative for S-100 protein and smooth muscle actin. There were many reports regarding early lesions of juvenile hyaline fibromatosis; however in this patient, tumor existed for more than 20 years and the histology was somewhat different from the early lesions reported in the literature.
- Multiple Plexiform Schwannomas Associated with Neurofibromatosis Type 2: A case report.
- Ho Sung Park, Myoung Ja Chung, Myoung Jae Kang, Dong Geun Lee, Byung Cook Ahn
- Korean J Pathol. 2000;34(5):389-392.
- 2,244 View
- 16 Download
-
Abstract
PDF
- Plexiform schwannoma is a rare benign tumor arising from the peripheral nerve sheath and characterized by a multinodular and plexiform growth pattern. This tumor usually arises sporadically. In rare cases, plexiform schwannomas have been associated with neurofibromatosis type 2. Plexiform schwannoma should be differentiated from plexiform neurofibroma, because the latter is pathognomonic tumor of neurofibromatosis type 1 and has a potential of malignant transformation. We report a case of multiple plexiform schwannomas associated with bilateral acoustic neuromas and meningioma.
- A Case of Desmoplastic Fibroma of the Mandible.
- Dong Won Kim, Tae Jung Kwon, Dong Wha Lee
- Korean J Pathol. 1988;22(3):340-347.
- 2,084 View
- 15 Download
-
Abstract
PDF
- A case of desmoplastic fibroma of mandible in a 18 years old woman is presented. She had complained progressive swelling of right mandible for 4 years. Radiographically, a multilocular radiolucent of right hemimandibulectomy showed multinodular external surface without cortical destruction. Cut surfaces revealed grayish white, fibrous homogeneous appearance with firm consistency, sparing head portion. The maximum diameter of the tumor was 13 cm. Microscopically, the tumor was composed of interlacing bundles of monomorphic spindle-shaped cells with abundant intercellular collagen. Ultrastructurally, most tumor cells were fibroblastic-like cells with abundant RER and cytoplasmic fibrils, but a few disclosed transition to myofibroblasts. However, no fully developed myofibroblasts were seen.
- Trichoblastic Fibroma: A Pathologic Analysis of 4 Cases.
- Ah Won Lee, Ji Han Jung, Jin Young Yoo, Seok Jin Kang, Byung Kee Kim
- Korean J Pathol. 2000;34(8):574-580.
- 2,829 View
- 54 Download
-
Abstract
PDF
- Trichoblastic fibroma is a benign trichogenic tumor that has both epithelial and mesenchymal components and exhibits partial follicular induction. We studied 4 cases of trichoblastic fibroma and reviewed their clinical and histologic features. Two tumors were present in the face. The remaining two were in the vulva and perianal area, respectively. The age of the patients ranged from 53 to 68 years, with an average age of 62. All were female. Histologically, the lesions showed a well circumscribed mass, located at dermo-subcutaneous junction in three patients and subcutaneous in one. They demonstrated mesenchymal induction evidenced by hair germ-like structure and perifollicular sheath. There was no connection between the tumor and epidermis. Differentiation toward hair structure led to the formation of the infundibulum through inner root sheath. Trichoblastic fibroma may be confused clinically and/or histologically with basal cell carcinoma. Identification of the mixed epithelial and mesenchymal components, and the absence of epidermal connection and cleft within the stroma are important in differentiating this benign neoplasm from basal cell carcinoma.
- Elastofibromatous Lesion of the Stomach: A case report.
- Mee Sook Roh, Sook Hee Hong
- Korean J Pathol. 1995;29(1):103-105.
- 2,340 View
- 34 Download
-
Abstract
PDF
- Elastofibroma is a peculiar tumor-like lesion which manifests as a slowly growing, solid, ill-defined mass of fibroelastic tissue occurring almost exclusively in elderly persons. It has been found in the ,,ubscapular region but rare examples have also been found in other locations. We experienced a case of elastofibromatous lesion of the stomach. The lesion was incidentally found in a 71 -year-old woman during an operation of cholecystectomy due to chronic cholecystitis and choledocholithiasis. The lesion was a relatively well-defined but not encapsulated small nodule, 0.7 cm in diameter, at submucosal layer of gastric pylorus. Histologically the nodular mass consisted of abundant acellular collagen fibers containing numerous elastofibroma fibers.
- Intraneural Perineurioma in the Tongue: A Case Report.
- Jun Kang, Shin Kwang Khang, Jene Choi, Jeong Won Kim, Eul Ju Seo, Bu kyu Lee, Eunsil Yu
- Korean J Pathol. 2007;41(1):51-54.
- 2,617 View
- 29 Download
-
Abstract
PDF
- We report a case of an intraneural perineurioma that developed in an unusual location, the tongue. A 16-year-old male presented with a 1 cm sized protruding submucosal mass in his tongue without any sensory or motor signs or symptoms. The mass was excised. The mucosa was intact, with an ill-defined firm mass measuring 1.0 x 0.8 x 0.6 cm in the submucosa and muscle. The cut surface of the mass was pinkish gray and fibrotic. Microscopically, the mass contained tortuous and thickened peripheral nerve bundles in the submucosa, showing onion bulb like structures. The onion bulb like structures consisted of centrally located S-100 protein positive Schwann cells surrounded by Glut-1 positive perineurial cells. The FISH study did not reveal any genetic aberrations in chromosome 22.
- Elastofibroma.
- Sang Yong Song, In Ae Park, Yong Il Kim
- Korean J Pathol. 1992;26(4):420-422.
- 2,108 View
- 10 Download
-
Abstract
PDF
- Elastofibroma is a rare benign tumorous growth presenting as a slowly growing ill-defined mass of fibroblastic tissue occurring in elderly persons and arising mainly form the connective tissue between the lower portion of the scapula and the chest wall. Its pathogenesis is not well established but it may be the result of nonneoplastic reactive hyperplasia taking place with constitutional predisposition in the background. A case of elastofibroma occurring in the subscapular area of a 65-year-old female cook is presented. The mass, 6x5x3 cm in maximum dimensions, was poorly circumscribed, solid, hard, pale fleshy and pray-white fibrous tumor. Microscopically, it was composed of numerous small globular and linear elastic fibers embedded in collagenous matrix. To our knowledge, it is the first case of elastofibroma in Korea.
- Clonal Analysis of Neurofibroma by PCR Amplification of HUMARA Gene.
- Jae Hyuk Lee, Seung Sang Han, Hyun Sik Oh, Yoo Duk Choi, Hyun Joong Kim, Kyung Hwa Lee, Jong Hee Nam, Chan Choi, Sang Woo Juhng
- Korean J Pathol. 2003;37(6):421-428.
- 2,187 View
- 14 Download
-
Abstract
PDF
- BACKGROUND
While neurofibromas have generally been regarded as polyclonal hyperplastic lesions, it remains unclear whether the tumor is a true neoplasm or a hyperplastic lesion.
METHODS
Determination of clonality by X chromosome inactivation pattern was investigated in twenty-one cases of neurofibroma employing enzyme digestion and PCR of the HUMARA gene. The histological, immunohistochemical, and ultrastructural characteristics of the tumors were also examined.
RESULTS
Immunohistochemically, most of the tumor cells showed vimentin and S-100 protein positivity. Axons were demonstrated by neurofilament protein positivity and were seen mainly at the periphery and rarely in the central portion of the tumor. Ultrastructurally, the tumors were composed of a variety of cell types: perineurial cells, Schwann cells, fibroblasts, and axons. X chromosome inactivation analysis was completed on thirteen out of fifteen cases in which DNA was successfully extracted. Of thirteen neurofibromas that were heterozygous at the HUMARA loci, eleven showed a polyclonal pattern. The remaining two cases were considered as indeterminate for clonality because of unequal band intensity and failure to obtain the normal control DNA.
CONCLUSION
The results from this study suggest that neurofibromas are polyclonal in origin and might be a neoplastic lesion comprising non-neoplastic cells among constituent components.
- Diffuse Neurofibromas: Clinicopathologic Analysis of 11 cases.
- So Young Park, Hye Kyung Lee, Se Min Baek
- Korean J Pathol. 1995;29(2):181-188.
- 2,349 View
- 20 Download
-
Abstract
PDF
- We reviewed surgical specimens from 11 patients with diffuse neurofibroma to define the specific clinicopathologic characteristics. Ten cases were cutaneous neurofibromas and one case was an uncommon gastrointestinal neurofibroma involving the rectum. The most frequent sites of involvement were the head and neck, especially the eyelids and the periorbital areas. They usually presented as a plaque-like elevation of the skin. They primarily occured in children and young adults and positive family histories of von Recklinghausen's neurofibromatosis were obtained in 45.4%. Pathologically, the involved skin & rectum were diffusely thickened by an infiltrative growing mass, showing proliferation of short fusiform cells in the uniform matrix of fine fibrillary collagen. The characteristic prominence of Wagner-Meissner bodies (45.4%) suggests they could be associated with pathogenesis of diffuse neurofibroma. On the basis of these findings, we could confirm diffuse neurofibroma to be a distinct form of neurofibroma.
- Osteofibrous Dysplasia-Like Adamantinoma: A Case Report with its Immunohistochemical and Ultrastructural Studies.
- Na Rae Kim, Geunghwan Ahn, Geun Woo Kim, Hyun Yee Cho, Young Ha Oh, Dong Hae Chung
- Korean J Pathol. 2004;38(1):50-55.
- 3,165 View
- 33 Download
-
Abstract
PDF
- Osteofibrous dysplasia (OFD)-like adamantinoma is a rare skeletal tumor that is characterized by the predominant OFD-like pattern with scattered epithelial nests. Adamantinoma shares clinical features (the majority of lesions in the tibia and the prevalent age group), radiologic findings (radiolucency with sclerotic shadow), and pathologic similarities (particularly the presence of scattered cytokeratin-positive stromal cells) with OFD. We describe a case of OFD-like adamantinoma. Epithelial cell nests express the epithelial membrane antigen, pancytokeratin, CK14, and collagen type IV. Ultrastructurally, the oval to spindle cells in the epithelial foci had abundant tonofilaments, and well-formed desmosomes with dense plaques, of which well preserved desmosomes are demonstrated for the first time in OFD-like adamantinoma. These immunohistochemical and ultrastructural findings further support that the origin of epithelial cells of classic and OFD-like adamantinoma are epithelial cells transformed from fibroblastic cells in the proliferating osteofibrous tissue.
 First
First Prev
Prev


