 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 57(1); 2023 > Article
-
Newsletter
What’s new in neuromuscular pathology 2022: myopathy updates and gene therapies -
Chunyu Cai

-
Journal of Pathology and Translational Medicine 2023;57(1):79-80.
DOI: https://doi.org/10.4132/jptm.2022.10.14
Published online: December 20, 2022
Department of Pathology, University of Texas Southwestern Medical Center, Dallas, TX, USA
- Corresponding Author: Chunyu Cai, MD, PhD, Department of Pathology, University of Texas Southwestern Medical Center, Dallas, Texas, USA, E-mail: Chunyu.Cai@utsouthwestern.edu
This article has been published jointly, with consent, in both Journal of Pathology and Translational Medicine and PathologyOutlines.com.
© 2023 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 5,138 Views
- 215 Download
Abstract
- This compilation of new changes in the diagnosis and treatment of muscle and nerve disease is extracted from the latest publications from the European Neuromuscular Centre International workshops, FDA.gov and clinicaltrials.gov.
- Classification of idiopathic inflammatory myopathies (IIM)
European Neuromuscular Centre (ENMC) clinico-sero-pathological classification divides IIM into 4 subclasses, which are associated with myositis specific antibodies (MSAs), as outlined in Table 1.
MSA testing is preferentially performed prior to immune suppression treatment but should also be performed in patients with suspected IIM or interstitial lung disease of unknown etiology without prior MSA testing.
Polymyositis no longer exists as an IIM subclass.
- Dermatomyositis (DM)
A classification of DM cannot be made in the absence of DM rash (Gottron’s sign, Gottron’s papules, heliotrope rash).
-
In the presence of DM-like rash, DM can be diagnosed if a DM-specific autoantibody is present, or if definitive DM muscle biopsy features are present and are combined with clinical signs of proximal muscle weakness or elevated muscle enzymes.
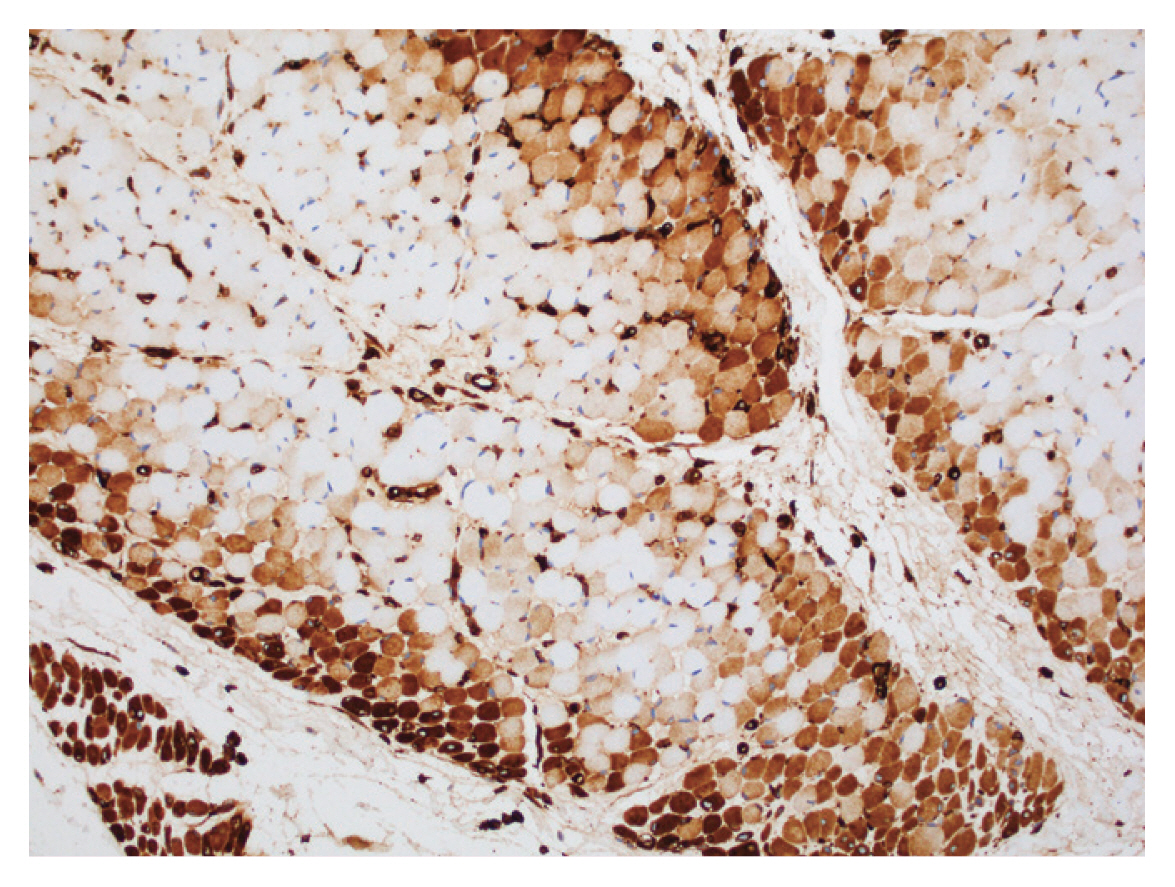
- Definitive DM muscle biopsy findings: perifascicular atrophy and/or perifascicular MxA overexpression (Fig. 1) with rare or absent perifascicular necrosis.
- Suggestive DM muscle biopsy findings: lymphocytic infiltrates (often perivascular), evidence of perifascicular disease (perifascicular predominant fibers that are pale on COX staining and/or positive on NCAM staining).
- DM specific autoantibodies: Mi2, NXP2, TIF1γ, MDA5, SAE. Patients are subclassified according to that autoantibody (e.g., anti-TIF1γ DM, anti-NXP2 DM). Patients who have DM without a DM-specific autoantibody are subclassified as having “autoantibody negative DM.”
- Antisynthetase syndrome (ASyS)
-
Defined by the presence of an antibody to aminoacyl tRNA synthetase (ARS), together with a single or combination of the following clinical manifestations:
ASyS patients with a DM-like rash are not classified as DM, but as “ASyS with a DM-like rash.”
-
Pathology is characterized by:
- Immune mediated necrotizing myopathies (IMNM)
Anti-HMGCR and anti-SRP autoantibodies are considered specific for IMNM.
Patients with anti-HMGCR antibody and a DM-like rash will be classified as having “anti-HMGCR myopathy with a DM-like rash,” while patients with anti-SRP antibody and a DM-like rash will be classified as having “anti-SRP myopathy with a DM-like rash.”
Pathology requires diffusely scattered necrotic fibers at different stages of resolution and macrophage dominant, pauci-lymphocytic inflammation.
- Sporadic inclusion body myositis (sIBM)
-
The pathologic criteria for sIBM include:
Anti-cN1A autoantibody is present in 30%–70% of sIBM patients but has also been found in DM and other systemic autoimmune diseases such as Sjögren’s and lupus.
Clinically, elderly patients with asymmetric muscle weakness and atrophy of proximal and distal muscle groups, with predilection for wrist and finger flexors and knee extensors.
Usually refractory to immunosuppressive therapies.
- Limb girdle muscular dystrophies (LGMD)
The definition and nomenclature of LGMD have been re-defined in the 2017 ENMC international workshop as a genetically inherited condition that primarily affects skeletal muscle leading to progressive, predominantly proximal muscle weakness at presentation caused by a loss of muscle fibers.
-
All LGMD subclasses must fulfill all of the following:
- Described in at least two unrelated families
- Patients have achieved independent walking (to differentiate from congenital muscular dystrophies)
- Elevated serum creatine kinase
- Degenerative changes on muscle imaging over the course of disease
- Dystrophic changes on muscle histology, ultimately leading to end-stage pathology
New nomenclature: change from the alphanumeric system to include the name of the affected protein and mode of inheritance (D for dominant, R for recessive, X for X-linked); examples listed in Table 2.
Some previous LGMD subclasses no longer fulfill the new LGMD definition (Table 3).
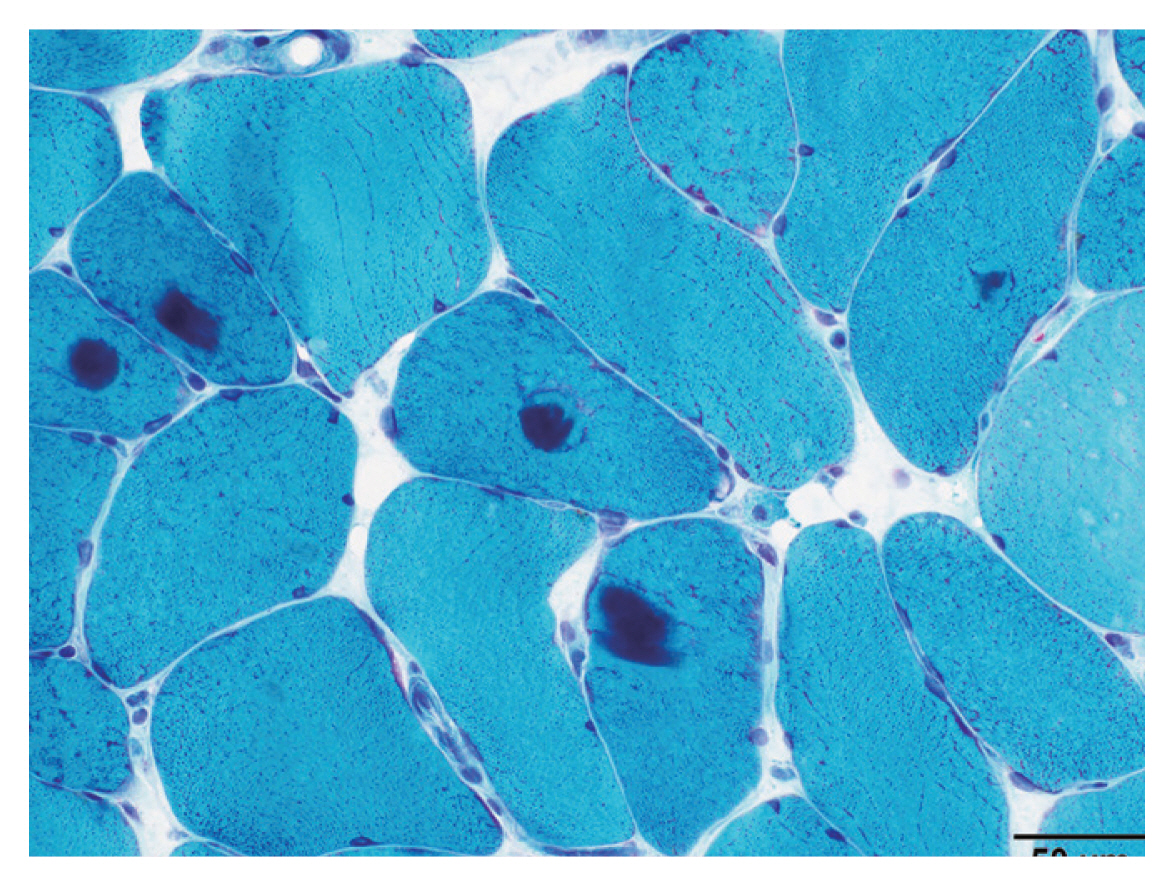
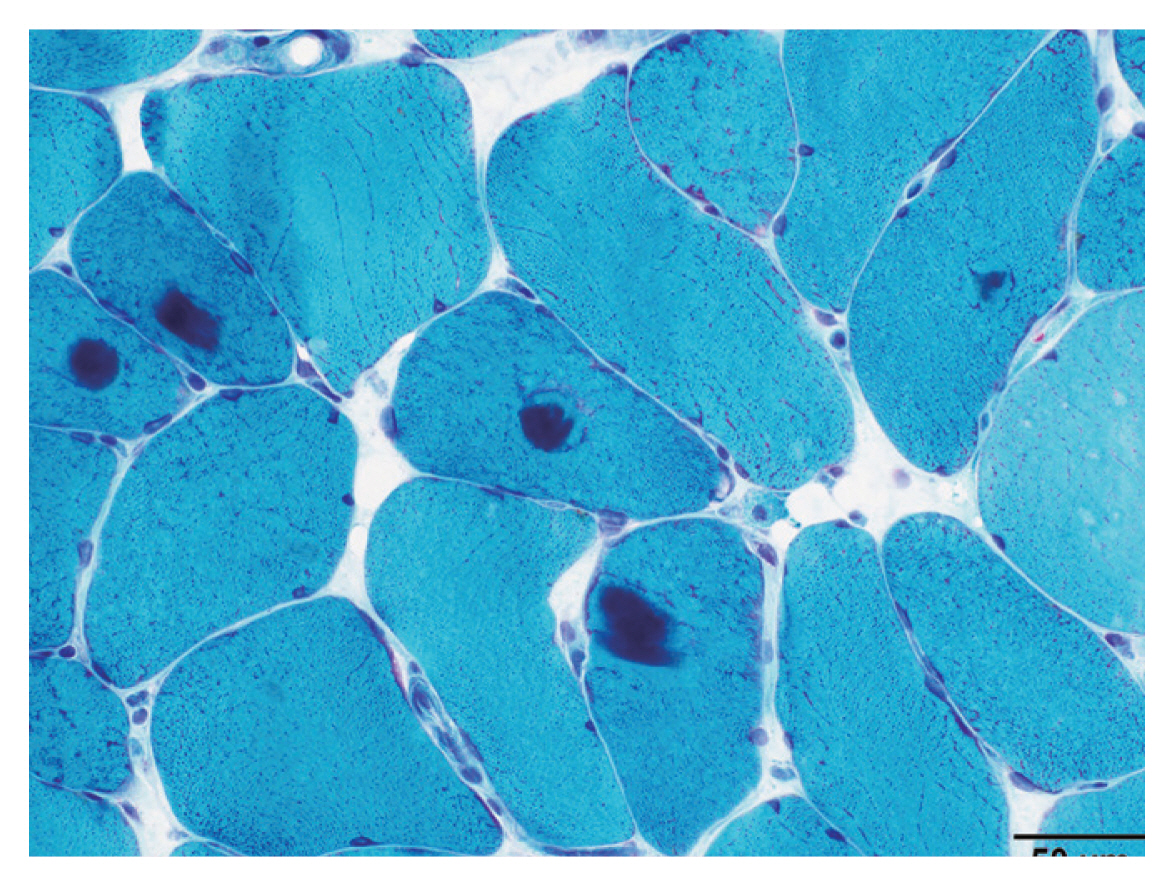
- Myofibrillar myopathies (MFM)
MFM is a group of disorders associated with myofibrillar degradation that begins in the Z disk.
Histologically characterized by sarcoplasmic pleomorphic amorphous, granular or hyaline protein aggregates on Gomori trichrome (Fig. 2), and positive for desmin immunostain.
Most contain mutations in Z disk associated protein coding genes: DES, CRYAB, MYOT, ZASP, FLNC, BAG3.
Patients with mutations in FHL1, DNAJB6, HSBP8, TTN, ACTA1, PLEC, and LMNA have also been associated with MFM phenotype.
- Myotonic dystrophy (DM1/DM2)
Autosomal dominant multi-system diseases with the common features of myotonia and progressive muscle weakness. There are two main forms: DM1 and DM2.
DM1 is caused by CTG trinucleotide repeats in the 3′ untranslated region of DMPK. DM1 shows striking anticipation, with age at onset decreasing by 20–30 years per generation.
DM1 muscle pathology is characterized by markedly increased internalized nuclei, often in chains and ring fibers, in a background of chronic myopathy.
DM2 is caused by CCTG repeat expansion in intron 1 of CNBP (ZNF9). Additionally, CLCN1 and SCN4A are disease modifying genes whose mutations may exaggerate DM2 phenotype; they therefore should be included in DM2 genetic screening.
DM2 muscle pathology is characterized by type 2 atrophy and frequent internalized nuclei predominantly in type 2 fibers.
MYOPATHY UPDATES
- Gene replacement therapies for hereditary neuromuscular diseases
Adeno-associated virus (AAV) based gene delivery vectors can produce replacement proteins in patients with loss of function mutations, such as spinal muscular atrophy (SMA) or Duchenne’s muscular dystrophy. The vector does not integrate into the patient’s genome and has a low immunogenicity.
CRISPR-Cas9-mediated gene editing does incorporate into the patient’s genome and can permanently replace a deleterious mutation in patient with conditions such as hereditary transthyretin-mediated (hATTR) amyloidosis.
Both methods entail only a single intravenous injection and thus have a clear advantage over siRNA based and antisense oligonucleotide (ASO) based therapies, which require serial infusions.
- FDA approved gene therapy for neuromuscular diseases
Zolgensma (Novartis) is the first ever FDA approved (2019), intravenously delivered, AAV9 vector mediated SMN gene therapy for spinal muscular atrophy.
- New gene therapies currently in clinical trials
NTLA-2001 is a CRISPR-Cas9-mediated gene editing construct that targets hATTR amyloidosis in a phase II–III trial for adults with polyneuropathy or cardiomyopathy (NCT04601051).
SRP-9001 (Sarepta), SGT-001 (Solid Biosciences) and PF-06939926 (Pfizer) are AAV based micro-dystrophin constructs in phase III trials for DMD (NCT03375164, NCT03769116, NCT04281485).
SPK-3006 (Spark Therapeutics) is an AAV based human GAA gene construct in a phase I/II trial for adult onset Pompe disease (NCT04093349).
- Meet the Author
- Dr. Chunyu Cai has been part of the PathologyOutlines.com editorial board and the Deputy Editor in Chief for Neuropathology since 2020. He is a pathologist and an Associate Professor at University of Texas Southwestern Medical Center. His research focuses on neuromuscular diseases and brain tumors.
GENE THERAPIES

| Dermatomyositis (DM) | Mi2, NXP2, TIF1γ, MDA5, SAE |
| Inclusion body myositis (IBM) | cN1A* |
| Immune mediated necrotizing myopathy (IMNM) | SRP, HMGCR |
| Anti-synthetase syndrome (ASys) | Jo-1, PL7, PL12, EJ, OJ, KS, Zo, Ha |
| Previous name | Gene | New name |
|---|---|---|
| LGMD 1D | DNAJB6 | LGMD D1 DNAJB6-related |
| LGMD 1I | CAPN | LGMD D4 Calpain3-related |
| LGMD 2A | CAPN | LGMD R1 Calpain3-related |
| LGMD 2B | DYSF | LGMD R2 Dysferlin-related |
Figure & Data
References
Citations
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
- Related articles
-
- What’s new in digital and computational pathology 2026: advances in adoption, standards, AI technologies, and clinical integration
- What's new in molecular genetic pathology 2026: emerging biomarkers for personalized cancer therapies
- What’s new in hematopathology 2025: myeloid neoplasms in the WHO 5th edition and ICC
- What’s new in medical renal pathology 2025: Updates on podocytopathy and immunofluorescence staining in medical kidney
- What’s new in neuropathology 2024: CNS WHO 5th edition updates


Fig. 1
Fig. 2
| Dermatomyositis (DM) | Mi2, NXP2, TIF1γ, MDA5, SAE |
| Inclusion body myositis (IBM) | cN1A |
| Immune mediated necrotizing myopathy (IMNM) | SRP, HMGCR |
| Anti-synthetase syndrome (ASys) | Jo-1, PL7, PL12, EJ, OJ, KS, Zo, Ha |
| Previous name | Gene | New name |
|---|---|---|
| LGMD 1D | DNAJB6 | LGMD D1 DNAJB6-related |
| LGMD 1I | CAPN | LGMD D4 Calpain3-related |
| LGMD 2A | CAPN | LGMD R1 Calpain3-related |
| LGMD 2B | DYSF | LGMD R2 Dysferlin-related |
| Previous name | Gene | New name |
|---|---|---|
| LGMD 1A | MYOT | Myofibrillar myopathy |
| LGMD 1B | LMNA | Emery–Dreifuss muscular dystrophy |
| LGMD 1C | CAV3 | Rippling muscle disease |
| LGMD 1E and 2R | DES | Myofibrillar myopathy |
| LGMD 1H | unknown | n/a |
| LGMD 2V | GAA | Pompe disease/acid maltase deficiency |
MSAs are usually mutually exclusive and specific for IIM subclasses, with the exception of cN1A
n/a, not available

