 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 49(1); 2015 > Article
-
Review
Genomic Landscapes of Pancreatic Neoplasia - Laura D. Wood, Ralph H. Hruban
-
Journal of Pathology and Translational Medicine 2015;49(1):13-22.
DOI: https://doi.org/10.4132/jptm.2014.12.26
Published online: January 15, 2015
The Sol Goldman Pancreatic Cancer Research Center, Johns Hopkins University School of Medicine, Baltimore, MD, USA
- Corresponding Author: Laura D. Wood, M.D. The Sol Goldman Pancreatic Cancer Research Center, Johns Hopkins University School of Medicine, CRB 2 Room 345, 1550 Orleans Street, Baltimore, MD 21231, USA Tel: +1-410-955-3511 Fax: +1-410-614-0671 E-mail: ldwood@jhmi.edu
© 2015 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- Abstract
- PANCREATIC DUCTAL ADENOCARCINOMA AND ITS VARIANTS
- FAMILIAL PANCREATIC CANCER GENES
- PRECURSORS TO PANCREATIC DUCTAL ADENOCARCINOMA: PANCREATIC INTRAEPITHELIAL NEOPLASIAS
- PRECURSORS TO PANCREATIC DUCTAL ADENOCARCINOMA: IPMNS AND MCNS
- OTHER CYSTIC NEOPLASMS
- SOLID-PSEUDOPAPILLARY NEOPLASMS
- PANCREATIC NEUROENDOCRINE TUMORS
- NEOPLASMS WITH ACINAR DIFFERENTIATION
- CONCLUSION
- NOTES
- Acknowledgments
- REFERENCES
Abstract
- Pancreatic cancer is a deadly disease with a dismal prognosis. However, recent advances in sequencing and bioinformatic technology have led to the systematic characterization of the genomes of all major tumor types in the pancreas. This characterization has revealed the unique genomic landscape of each tumor type. This knowledge will pave the way for improved diagnostic and therapeutic approaches to pancreatic tumors that take advantage of the genetic alterations in these neoplasms.
- Pancreatic ductal adenocarcinoma (PDAC) is the most common pancreatic malignancy, and with a 5-year survival of only 6%, it is one of the deadliest of all human cancers. The KRAS gene, which encodes a small GTPase that mediates downstream signaling from growth factor receptors, is the most commonly mutated oncogene in PDAC [1-3]. Missense mutations in KRAS cluster in specific hotspots (most commonly codon 12), consistent with its role as an oncogene—KRAS mutations occur in >90% of PDACs. In addition to hotspot mutations in the KRAS oncogene, three tumor suppressor genes are frequently mutated in PDAC. The tumor suppressor gene P16/CDKN2A, which encodes a critical cell cycle regulator, is inactivated in >90% of PDACs by several mechanisms, including intragenic mutation coupled with loss of heterozygosity, homozygous deletion, and promoter methylation [4]. Mutations in TP53, a key component of the cellular stress response, are also common in PDAC, reported in approximately 75% of PDACs, most commonly by small intragenic mutation coupled with loss of heterozygosity [5,6]. Mutations in TP53 often result in strong diffuse nuclear expression of p53 protein which can be detected by immunohistochemistry (Fig. 1A). Somatic inactivation of SMAD4, a tumor suppressor gene that codes for a component of the transforming growth factor beta signaling pathway, occurs in approximately 55% of PDACs, usually by homozygous deletion or intragenic mutation coupled with loss of heterozygosity [7,8]. These mutations can also be detected by immunohistochemistry, as they cause loss of Smad4 protein expression (Fig. 1B). Mutations in SMAD4 have potential clinical implications, as these mutations are associated with worse prognosis and widespread metastases (rather than local disease) [9,10]. Although these four genes (often referred to as the four “mountains”) are the most commonly mutated genes in pancreatic cancer (Table 1), only a minority of patients (37% in one study) have mutations in all four genes, highlighting the genetic heterogeneity of the disease [11].
- Mutations in these four driver genes were well described before the introduction of high throughput sequencing. However, multiple studies have now examined the whole exomes and whole genomes of large numbers of PDACs, and these studies have deepened our understanding of the pancreatic cancer in many ways [6,12]. First, these studies confirmed the importance of the four key driver genes (KRAS, P16/CDKN2A, TP53, and SMAD4) as the most frequently altered genes in PDAC. These studies also identified numerous other genes that are less commonly somatically mutated in PDACs—the average number of nonsynonymous genetic alterations ranged from 26 to 63 in the two key studies [6,12]. Several of these genes have known roles in tumorigenesis, including MLL3, TGFBR2, ATM, and ARID1A, and thus are likely to be drivers in pancreatic cancer in spite of their low mutation rate. However, many of the other infrequently mutated genes have no known role in cancer, and thus it is not possible to separate out driver mutations (which have a functional effect on tumorigenesis) from passenger mutations (which have no functional effect but instead accumulate randomly through repeated rounds of cell division within a tumor). Overall, these data highlight the heterogeneity at the gene level among different PDACs from different patients. However, although the individual genes altered are markedly heterogeneous, PDACs share many similarities when mutated genes are not considered at the individual gene level, but instead are considered as components of their larger biological pathways: there are several core processes and pathways that are genetically altered in the majority of carcinomas sequenced. These commonly altered pathways, which include KRAS signaling, DNA damage control, and cell adhesion, represent shared features of pancreatic tumorigenesis. In addition to these pathways with clear links to processes critical to tumor formation, one study also identified frequent mutations in genes in the axon guidance pathway as promising potential drivers in PDAC [12].
- Somatic mutations have also been used to understand metastasis and model the time course of pancreatic tumorigenesis. Studies of somatic mutations in matched primary tumors and metastases did not identify any genetic alterations that were specific to metastasis [13]. Instead, the vast majority of the mutations identified in metastases were present in a subclonal population of the primary tumor. Modeling based on these data suggests a time period of approximately 15 years between the occurrence of the initiating mutation in PDAC and the acquisition of metastatic ability [13]. These data are encouraging, as they suggest a broad time window for early detection of PDAC while it is still curable by surgery [14].
- Although the contribution of protein-altering somatic mutations is well documented in PDAC, it is likely that other genetic and epigenetic alterations also play a role in pancreatic tumorigenesis. This idea is supported by the observation that copy number alterations and promoter methylation affect known drivers of PDAC [15]. Many studies have identified copy number gains and losses as well as complex karyotypes in PDAC, and several studies have examined differential expression of micro-RNAs [16-21]. However, because of difficulties in determining the target genes and functional effects of these types of alterations, it is much more challenging to separate out driver and passenger alterations when analyzing large chromosomal alterations as well as differences in gene and microRNA expression.
- There are several uncommon variants of PDAC—most of these are genetically similar to PDAC with respect to mutations in key driver genes (KRAS, P16/CDKN2A, TP53, and SMAD4). However, some variants also have unique genetic alterations. For example, in addition to mutations in previously known drivers, adenosquamous carcinoma has frequent somatic mutations in UPF1, which encodes a crucial component of the RNA degradation pathway of nonsense mediated decay [22,23]. The mutations in UPF1 cause aberrant splicing of the mRNA, eliminating essential protein domains and potentially conferring dominant negative activity [22]. Colloid carcinomas, which are characterized by large pools of stromal mucin and are associated with intestinal-type intraductal papillary mucinous neoplasms (IPMNs, see below), have somatic mutations in GNAS (which are not typically seen in PDAC), as well as KRAS and TP53 [24]. Medullary carcinomas exhibit a high prevalence of microsatellite instability [25,26], and undifferentiated carcinomas show frequent loss of E-cadherin protein expression [27] (Fig. 1C). Although no somatic mutations in CDH1 have been reported in undifferentiated carcinomas, promoter methylation has been reported and may explain the loss of E-cadherin protein expression [27].
PANCREATIC DUCTAL ADENOCARCINOMA AND ITS VARIANTS
- A number of genes have been identified that increase the risk of developing PDAC when altered in the germline (Table 2). These genes are important to recognize for three reasons. First, the risk of developing pancreatic cancer can be quantified when the gene is known; this knowledge can help guide patient care and research screening. For example, germline mutations in PRSS1 cause chronic pancreatitis and greatly increase the risk of pancreatic cancer [28]. Some of these individuals, particularly those with a nonfunctioning pancreas from years of chronic pancreatitis, choose prophylactic pancreatectomy to reduce their risk of developing PDAC [29]. Second, the risk of developing extra-pancreatic malignancies can be quantified when the gene is known. For example, germline P16/CDN2KA mutations increase the risk of both PDAC and melanoma and lives can be saved by screening carriers of a germline P16/CDN2KA mutation and their biological relatives for melanoma [30,31]. Third, some of the germline mutations result in changes in the PDACs that are potentially therapeutically targetable. For example, germline BRCA2 mutations increase the risk of ovarian, breast and pancreatic cancer, and the pancreatic cancers that arise in patients with a germline BRCA2 mutation may be particularly sensitive to poly (ADP-ribose) polymerase (PARP) inhibitors and to DNA cross-linking agents such as cisplatinum [32,33].
FAMILIAL PANCREATIC CANCER GENES
- PDACs arise from histologically well-defined precursor lesions. The majority of PDACs arise from pancreatic intraepithelial neoplasia (PanIN), which are microscopic intraductal lesions and too small to be detected using currently available imaging technologies. However, a significant minority of PDACs arise from cystic precursors that can be identified with currently available imaging technologies—these cystic precursor lesions include IPMNs and mucinous cystic neoplasms (MCNs). Although some alterations are shared among these precursor lesions, there are genetic features that distinguish PanINs, IPMNs, and MCNs.
- PanINs, which are categorized based on the morphological grade of dysplasia as low-grade (PanIN-1 and PanIN-2) and highgrade (PanIN-3), sequentially acquire the driver gene mutations that characterize PDAC. KRAS mutations are an early event in PanIN formation, occurring in >90% of even the lowest grade PanINs [34]. P16/CDKN2A mutations also occur in PanIN-1, though far less frequently than KRAS mutations, and the prevelance of P16/CDKN2A mutations increase with PanIN grade [34]. However, loss of Smad4 expression and TP53 alterations are late events. Smad4 loss has been reported only in high-grade PanIN (PanIN-3) and invasive carcinoma—the same pattern has been reported for TP53 alterations [35,36]. In addition to mutations in these driver genes, telomere shortening is a very early event in PanINs, occurring in approximately 90% of PanIN-1s [37]. More extensive genetic characterization of PanINs is complicated by several factors. First, these lesions are small and thus only yield enough DNA for specific targeted genetic analyses. Second, as these lesions are only visible microscopically, it is very difficult to prospectively identify them and harvest fresh tissue; thus, the vast majority of PanIN lesions are only available from formalinfixed paraffin-embedded tissue. The study of high-grade PanIN (PanIN-3) is particularly difficult. Because PDAC can invade into and then spread along the duct system, it can be impossible to distinguish true PanIN-3 from the intraductal spread of invasive carcinoma. In spite of these difficulties, one study has reported whole exome sequencing of PDAC and associated PanINs [38]. This study found that approximately two-thirds of the somatic mutations were shared between PanINs and associated invasive carcinoma, while 10% were unique to the invasive carcinoma and 25% were unique to the PanINs. This study confirms the genetic relationship between PanINs and invasive carcinoma but also suggests additional genetic evolution with clinical progression of the disease.
PRECURSORS TO PANCREATIC DUCTAL ADENOCARCINOMA: PANCREATIC INTRAEPITHELIAL NEOPLASIAS
- Cystic pancreatic cancer precursors, IPMNs and MCNs, can also give rise to PDAC. Both are cystic lesions but have distinct clinical and morphological differences. IPMNs are more common in the head of the pancreas and involve the pancreatic duct system. MCNs are more common in the tail of the pancreas, occur almost exclusively in women, do not involve the duct system, and have a characteristic ovarian-type stroma. Like PanINs, IPMNs and MCNs are also classified by their grade of dysplasia (low, intermediate, or high). In addition, IPMNs are also classified by the direction of differentiation of the neoplastic epithelium into gastric, intestinal, pancreatobiliary, and oncocytic subtypes.
- IPMNs share mutations in the four key drivers of invasive PDAC (Table 1). KRAS mutations are prevalent in IPMNs and are present in up to 80% of tumors, occurring even in IPMNs with low-grade dysplasia [24,39]. Loss of p16 expression occurs in both noninvasive IPMNs and in IPMN associated invasive carcinomas, but it is much more common in invasive carcinomas (100% of invasive carcinomas vs 10% of noninvasive IPMNs in one study) [40]. Similarly, Smad4 loss is rare in noninvasive IPMNs but occurs in about one-third of IPMN-associated invasive carcinomas, identifying this as a late alteration in IPMN associated tumorigenesis [40,41]. TP53 mutations and protein overexpression are also limited to areas of high-grade dysplasia or invasive carcinoma in IPMNs [42-45].
- In addition to these drivers shared with conventional PDAC, IPMNs also have alterations in genes not typically targeted in PDAC (Table 1). Approximately 60% of IPMNs have somatic mutations in the oncogenic hotspot of GNAS, which encodes a protein that couples transmembrane receptors to their downstream signaling proteins [24]. These mutations occur in all grades of dysplasia and are most prevalent in intestinal-type IPMNs [39,46]. Other genetic alterations that underlie the morphological differences between the IPMN subtypes remain unknown. In addition, inactivating mutations in RNF43, a gene coding for an ubiquitin ligase, occur in approximately 60% of IPMNs [47]. The mutations in GNAS and RNF43 are striking in their prevalence as well as mutation types. The mutations in GNAS cluster at a single oncogenic hotspot codon, unequivocally establishing this gene as an oncogene. In contrast, the mutations in RNF43 are striking enriched for nonsense and other inactivating mutations, indicating that this is a tumor suppressor gene. Other potential driver genes that are mutated in IPMN but not PDAC include PIK3CA (10% of IPMNs) and STK11/LKB1 (5% of IPMNs) [48,49]. Whole exome sequencing of IPMNs revealed an average of 26 nonsynonymous somatic mutations per tumor, far fewer than in PDAC [47].
- Like IPMNs, MCNs share mutations in the four key drivers of PDAC—KRAS mutation is an early event, while tumor suppressor gene mutations (TP53, SMAD4, P16/CDKN2A) occur in MCNs with high-grade dysplasia or associated invasive carcinomas (Table 1). However, unlike IPMNs, GNAS mutations have not been identified in MCNs. Inactivating RNF43 mutations do occur in MCNs, but at a lower prevalence than in IPMNs [47]. Whole exome sequencing revealed an average of 16 nonsynonymous somatic mutations per IPMN [47].
PRECURSORS TO PANCREATIC DUCTAL ADENOCARCINOMA: IPMNS AND MCNS
- In addition to the cystic precursors to pancreatic cancer (IPMNs and MCNs), serous cystadenomas (SCAs) are clinically, pathologically, and genetically distinct from IPMNs and MCNs. SCAs are important to recognize because they are benign neoplasms with negligible risk of malignant behavior—these neoplasms are only resected if they cause symptoms due to mass effect or if there is clinical/radiologic concern for an IPMN. They are not precursors to invasive cancer. Whole exome sequencing of SCAs revealed an average of only 10 nonsynonymous somatic mutations per tumor, far fewer than in PDAC or its cystic precursors [47]. Only VHL was identified as a definite tumor suppressor gene in this tumor type, with somatic mutation in 50% and loss of heterozygosity in 90% of tumors (Table 1) [47]. SCAs occur as part of the clinical syndrome in von Hippel–Lindau syndrome, which results in clear cell neoplasms in various organs and is caused by germline mutations in VHL on chromosome 3—as such, the occurrence of somatic VHL mutations in sporadic SCAs is not surprising. Taken together, these data support the assertion that VHL is the major tumor suppressor responsible for the formation of both familial and sporadic SCAs. In addition, recent data show that vascular endothelial growth factor, a downstream target of VHL, is a marker of SCAs in pancreatic cyst fluid [50]. No mutations have been identified in PDAC or IPMN/MCN driver genes in SCAs. In addition to frequent loss of heterozygosity on chromosome 3p (the VHL locus), allelic loss of chromosome 10q has also been reported in 50% of cases and losses in several other chromosomes have been reported in multiple SCAs; however, no target genes for these losses has been identified [51].
OTHER CYSTIC NEOPLASMS
- Solid-pseudopapillary neoplasms (SPNs) are uncommon pancreatic neoplasms with low malignant potential—although surgical resection is curative for most patients, metastases can rarely occur [52,53]. Activating somatic mutations in CTNNB1 occur in almost all cases, leading to abnormal nuclear accumulation of β-catenin which can be detected with immunohistochemistry (Fig. 1D) [54,55]. Importantly, this nuclear accumulation of β-catenin on immunohistochemical stains can be used to support the histological diagnosis of SPN. Whole exome sequencing of SPNs revealed shockingly few somatic mutations, an average of only three nonsynonymous somatic mutations per tumor [47]. Some tumors in this sequencing study had only one somatic mutation, but all SPNs had a somatic mutation in CTNNB1. This demonstrates a unique feature of SPNs: a remarkably low number of somatic mutations, lower than any tumor type described to date, including pediatric tumors. Additional studies are required to determine whether tumorigenesis in this tumor type is driven by other types of alterations, such as large chromosomal translocations or epigenetic events.
SOLID-PSEUDOPAPILLARY NEOPLASMS
- Pancreatic neuroendocrine tumors (PanNETs), also known as islet cell tumors, are a distinct pancreatic neoplasm with neuroendocrine differentiation, as demonstrated by morphology or immunohistochemistry. They are classified as “syndromic” if they express hormones that result in a clinical syndrome, and as “familial” if they arise in a patient with a genetic predisposition. PanNETs are graded based on their proliferation rate, as assessed by mitotic count or Ki67 labeling index [56]. Tumors with >20 mitoses per high power field or Ki67 index >20% are considered neuroendocrine carcinomas, and neuroendocrine carcinomas can be further subdivided into large cell and small cell carcinomas.
- Whole exome sequencing of sporadic low-grade PanNETs identified an average of only 16 nonsynonymous somatic mutations per tumor [57]. Several driver genes unique to this tumor type were identified (Table 1). Somatic mutations in the MEN1 gene were identified in 45% of sporadic PanNETs [57,58]. Considering PanNETs are a major clinical manifestation of multiple endocrine neoplasia type 1 syndrome caused by germline mutation in MEN1, the presence of somatic mutations in this gene in sporadic PanNETs is not surprising. In addition, mutually exclusive somatic mutations in the chromatin remodeling genes ATRX and DAXX were identified in 45% of sporadic PanNETs [57]. These genes, which were enriched for inactivating mutations in PanNETs, function as part of a complex that is important for telomere maintenance—inactivation of ATRX and DAXX is associated with the telomerase independent telomere maintenance mechanism known as alternative lengthening of telomeres [59]. Studies in patients with MEN1 syndrome identified loss of ATRX and DAXX expression only in large PanNETs, suggesting that alterations in these genes are late events in pancreatic neuroendocrine tumorigenesis [60]. Somatic mutations in genes in the mammalian target of rapamycin cell signaling pathway (including PIK3CA, PTEN, and TSC2) occur in approximately 15% of sporadic PanNETs [57]. These alterations carry clinical significance, as drugs targeting this pathway have been developed for clinical use, and one such drug (Everolimus) has shown promise in PanNETs [61]. PanNETs lack frequent genetic alterations in major driver genes of PDAC—rare TP53 mutations have been reported, but no mutations in KRAS or P16/CDKN2A have been reported [57,62]. However, P16/CDKN2A promoter hypermethylation has been reported to be frequent in gastrinomas [62]. Although SMAD4 mutations were reported in one small study of nonfunctional PanNETs, this finding was not replicated in the whole exome sequencing study of PanNETs [57,63].
- Intriguingly, high-grade neuroendocrine carcinomas are genetically distinct from low-grade PanNETs. Neuroendocrine carcinomas retain expression of ATRX and DAXX and instead have alterations of P53 and RB1 [64].
PANCREATIC NEUROENDOCRINE TUMORS
- Two rare neoplasms of the pancreas exhibit acinar differentiation: acinar cell carincoma and pancreatoblastoma. While acinar cell carcinoma typically occurs in adults, pancreatoblastoma is more common in children and can occur in patients with Beck-with-Wiedemann syndrome, a disorder associated with imprinting dysregulation of chromosome 11p leading to overgrowth of various organs and predisposition to embryonal tumors [65].
- Acinar cell carcinomas are characterized by striking genomic instability at both the base pair and chromosomal level. In a recent whole exome sequencing study, each carcinoma had an average of 119 nonsynonymous somatic mutations, a larger number than any other pancreatic neoplasm [66]. Approximately 10% of acinar cell carincomas had microsatellite instability and a very large number of somatic mutations. Chromosomal instability was demonstrated by high fractional allelic losses in acinar cell carcinomas—sites of frequent loss included chromosome 11p (which was previously reported to be lost in a large proportion of acinar cell carcinomas) as well as tumor suppressor loci on chromosome 17p (TP53) and 18q (SMAD4) [67]. There was also a striking diversity in the genes altered by small somatic mutations in the whole exome sequencing—no single gene was altered in more than 30% of the acinar cell carcinomas sequenced. Mutations were identified in genes known to be drivers of PDAC (SMAD4 and TP53), cystic neoplasms (GNAS and RNF43), and PanNET (MEN1), as well as genes known to be drivers in other extrapancreatic tumor types. The mutations identified in APC and CTNNB1 confirmed previously reported findings of the importance of the Wnt signaling pathway in acinar cell carcinomas [67]. Other intriguing driver gene mutations identified in small subsets of acinar cell carcinomas included JAK1, BRAF, RB1, PTEN, ARID1A, MLL3, and BAP1. Importantly, these data suggest that therapies targeting mutations in JAK1 and BRAF could show promise in patients with acinar cell carcinoma. Many other genes were also mutated in 10%–20% of acinar cell carcinomas in the whole exome sequencing study, but further functional studies are required to determine the role of these mutations (if any) in tumorigenesis. Recent genetic studies have also identified frequent genomic rearrangements involving RAF genes (BRAF and RAF1) in almost one-fourth of acinar cell carcinomas [68]. Functional studies of the most frequently formed fusion gene SND1-BRAF show that it activates the mitogen-activated protein kinase pathway and confers sensitivity to MEK inhibition, pointing to a promising targeted therapy for a subset of patients with acinar cell carcinoma [68]. This study also confirmed the observation in the whole exome sequencing data of frequent mutations in DNA repair genes, suggesting potential utility for platinum-based chemotherapy or PARP inhibitors.
- Loss of chromosome 11p is the most frequent genetic alteration in pancreatoblastomas, occurring in >80% of tumors [69]. Like acinar cell carcinomas, pancreatoblastomas also have mutations in Wnt pathway genes: activating CTNNB1 mutations are more common than inactivating APC mutations in pancreatoblastoma, although both can occur [69]. Pancreatoblastomas lack frequent mutations in the key PDAC driver genes. Although at least a subset of pancreatoblastomas contain large chromosomal alterations, these alterations are difficult to interpret since no target genes in these alterations have been identified [70-73]. Recent whole exome sequencing studies included two pancreatoblastomas, and although the number of tumors sequenced is small, a few observations can be made [66]. First, these tumors contained far fewer mutations than the acinar cell carcinomas—17 and 18 nonsynonymous somatic mutations per pancreatoblastoma. Second, both pancreatoblastomas had somatic mutations in CTNNB1, supporting previous studies demonstrating the importance of Wnt pathway alterations in this tumor type.
NEOPLASMS WITH ACINAR DIFFERENTIATION
- Although the genetic alterations underlying pancreatic neoplasms have been studied for decades, the recent development of high throughput sequencing has enabled systematic characterization of the genomes of all the major tumor types in the pancreas. These studies have confirmed previously identified drivers of pancreatic neoplasia as well as identified previously unknown genes that likely play a crucial role in tumorigenesis. Each type of pancreatic neoplasm has a unique genetic profile, and many potential therapeutic targets have been identified. As we enter into the era of genomic medicine, knowledge of the molecular alterations in pancreatic neoplasms will become a critical component of clinical care, as these alterations will likely form the basis for early detection strategies and targeted therapeutic approaches. This has the potential to lead to great improvements in the lives of patients with pancreatic neoplasms, as diagnostic and therapeutic approaches will be targeted to an individual patient’s tumor.
CONCLUSION
Conflicts of Interest
Dr. Hruban receives royalty payments from Myriad Genetics for the PALB2 invention.
Acknowledgments
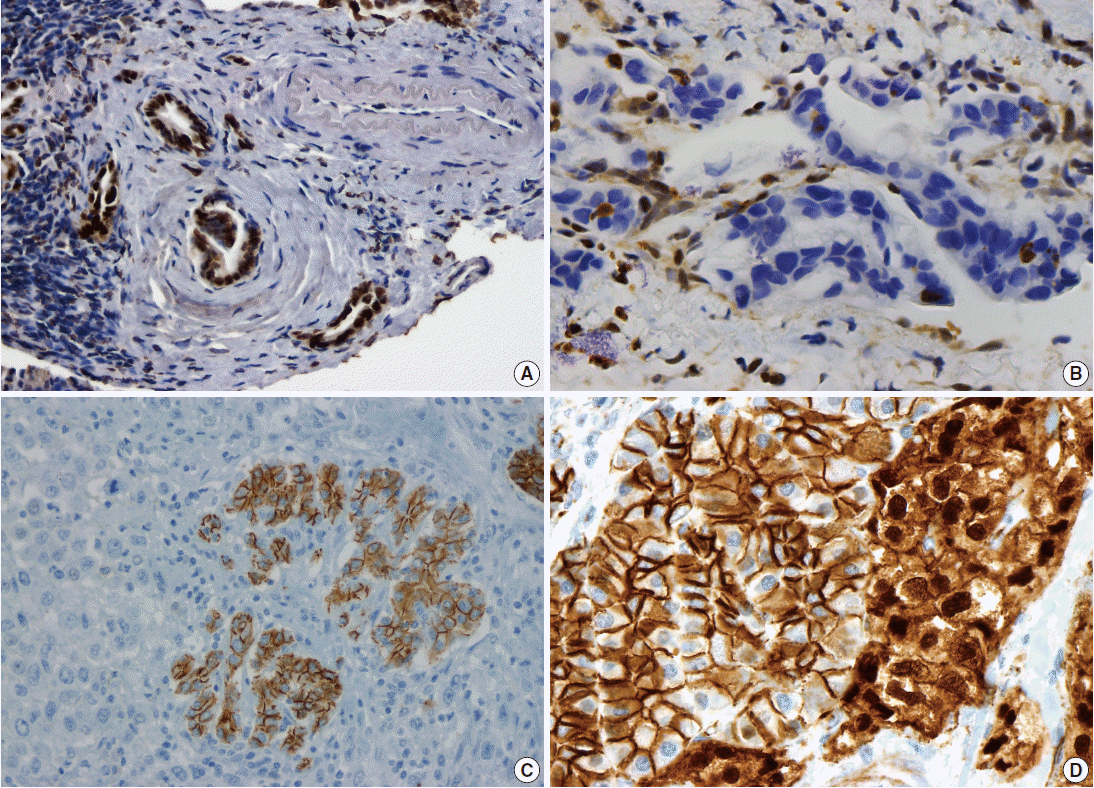
PDAC, pancreatic ductal adenocarcinoma; IPMN, intraductal papillary mucinous neoplasm; HGD, high-grade dysplasia; MCN, mucinous cystic neoplasm; carcinoma, invasive carcinoma; SCA, serous cystadenoma; SPN, solid-pseudopapillary neoplasm; PanNET, well-differentiated pancreatic neuroendocrine tumor; mTOR, mammalian target of rapamycin; ACC, acinar cell carcinoma; PB, pancreatoblastoma.
- 1. Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988; 53: 549-54. ArticlePubMed
- 2. Smit VT, Boot AJ, Smits AM, Fleuren GJ, Cornelisse CJ, Bos JL. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res 1988; 16: 7773-82. ArticlePubMedPMC
- 3. Hruban RH, van Mansfeld AD, Offerhaus GJ, et al. K-ras oncogene activation in adenocarcinoma of the human pancreas: a study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am J Pathol 1993; 143: 545-54. PubMedPMC
- 4. Schutte M, Hruban RH, Geradts J, et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res 1997; 57: 3126-30. PubMed
- 5. Rozenblum E, Schutte M, Goggins M, et al. Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res 1997; 57: 1731-4. PubMed
- 6. Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008; 321: 1801-6. ArticlePubMedPMC
- 7. Iacobuzio-Donahue CA, Song J, Parmiagiani G, Yeo CJ, Hruban RH, Kern SE. Missense mutations of MADH4: characterization of the mutational hot spot and functional consequences in human tumors. Clin Cancer Res 2004; 10: 1597-604. PubMed
- 8. Wilentz RE, Su GH, Dai JL, et al. Immunohistochemical labeling for dpc4 mirrors genetic status in pancreatic adenocarcinomas: a new marker of DPC4 inactivation. Am J Pathol 2000; 156: 37-43. PubMedPMC
- 9. Blackford A, Serrano OK, Wolfgang CL, et al. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin Cancer Res 2009; 15: 4674-9. ArticlePubMedPMCPDF
- 10. Iacobuzio-Donahue CA, Fu B, Yachida S, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol 2009; 27: 1806-13. ArticlePubMedPMC
- 11. Yachida S, White CM, Naito Y, et al. Clinical significance of the genetic landscape of pancreatic cancer and implications for identification of potential long-term survivors. Clin Cancer Res 2012; 18: 6339-47. ArticlePubMedPMCPDF
- 12. Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012; 491: 399-405. PubMedPMC
- 13. Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010; 467: 1114-7. ArticlePubMedPMCPDF
- 14. Lennon AM, Wolfgang CL, Canto MI, et al. The early detection of pancreatic cancer: what will it take to diagnose and treat curable pancreatic neoplasia? Cancer Res 2014; 74: 3381-9. ArticlePubMedPMCPDF
- 15. Sato N, Fukushima N, Maitra A, et al. Discovery of novel targets for aberrant methylation in pancreatic carcinoma using high-through-put microarrays. Cancer Res 2003; 63: 3735-42. PubMed
- 16. Bloomston M, Frankel WL, Petrocca F, et al. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA 2007; 297: 1901-8. ArticlePubMed
- 17. Lee EJ, Gusev Y, Jiang J, et al. Expression profiling identifies micro-RNA signature in pancreatic cancer. Int J Cancer 2007; 120: 1046-54. ArticlePubMedPMC
- 18. Szafranska AE, Davison TS, John J, et al. MicroRNA expression alterations are linked to tumorigenesis and non-neoplastic processes in pancreatic ductal adenocarcinoma. Oncogene 2007; 26: 4442-52. ArticlePubMedPDF
- 19. Griffin CA, Hruban RH, Morsberger LA, et al. Consistent chromosome abnormalities in adenocarcinoma of the pancreas. Cancer Res 1995; 55: 2394-9. ArticlePubMed
- 20. Iacobuzio-Donahue CA, van der Heijden MS, Baumgartner MR, et al. Large-scale allelotype of pancreaticobiliary carcinoma provides quantitative estimates of genome-wide allelic loss. Cancer Res 2004; 64: 871-5. ArticlePubMedPDF
- 21. Birnbaum DJ, Adélaïde J, Mamessier E, et al. Genome profiling of pancreatic adenocarcinoma. Genes Chromosomes Cancer 2011; 50: 456-65. ArticlePubMed
- 22. Liu C, Karam R, Zhou Y, et al. The UPF1 RNA surveillance gene is commonly mutated in pancreatic adenosquamous carcinoma. Nat Med 2014; 20: 596-8. ArticlePubMedPMCPDF
- 23. Brody JR, Costantino CL, Potoczek M, et al. Adenosquamous carcinoma of the pancreas harbors KRAS2, DPC4 and TP53 molecular alterations similar to pancreatic ductal adenocarcinoma. Mod Pathol 2009; 22: 651-9. ArticlePubMedPDF
- 24. Wu J, Matthaei H, Maitra A, et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med 2011; 3: 92ra66.ArticlePubMedPMC
- 25. Wilentz RE, Goggins M, Redston M, et al. Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas: a newly described and characterized entity. Am J Pathol 2000; 156: 1641-51. PubMedPMC
- 26. Goggins M, Offerhaus GJ, Hilgers W, et al. Pancreatic adenocarcinomas with DNA replication errors (RER+) are associated with wildtype K-ras and characteristic histopathology. Poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. Am J Pathol 1998; 152: 1501-7. PubMedPMC
- 27. Winter JM, Ting AH, Vilardell F, et al. Absence of E-cadherin expression distinguishes noncohesive from cohesive pancreatic cancer. Clin Cancer Res 2008; 14: 412-8. ArticlePubMedPMCPDF
- 28. Whitcomb D, Greer J. Germ-line mutations, pancreatic inflammation, and pancreatic cancer. Clin Gastroenterol Hepatol 2009; 7(11 Suppl): S29-34. ArticlePubMed
- 29. Bellin MD, Freeman ML, Gelrud A, et al. Total pancreatectomy and islet autotransplantation in chronic pancreatitis: recommendations from PancreasFest. Pancreatology 2014; 14: 27-35. ArticlePubMed
- 30. van der Rhee JI, Boonk SE, Putter H, et al. Surveillance of second-degree relatives from melanoma families with a CDKN2A germline mutation. Cancer Epidemiol Biomarkers Prev 2013; 22: 1771-7. ArticlePubMedPDF
- 31. Lynch HT, Fusaro RM, Lynch JF, Brand R. Pancreatic cancer and the FAMMM syndrome. Fam Cancer 2008; 7: 103-12. ArticlePubMedPDF
- 32. Lowery MA, Kelsen DP, Stadler ZK, et al. An emerging entity: pancreatic adenocarcinoma associated with a known BRCA mutation: clinical descriptors, treatment implications, and future directions. Oncologist 2011; 16: 1397-402. ArticlePubMedPMCPDF
- 33. O’Sullivan CC, Moon DH, Kohn EC, Lee JM. Beyond breast and ovarian cancers: PARP inhibitors for BRCA mutation-associated and BRCA-like solid tumors. Front Oncol 2014; 4: 42.PubMedPMC
- 34. Kanda M, Matthaei H, Wu J. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012; 142: 730-3.e9. ArticlePubMedPMC
- 35. Wilentz RE, Iacobuzio-Donahue CA, Argani P, et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res 2000; 60: 2002-6. PubMed
- 36. Maitra A, Adsay NV, Argani P, et al. Multicomponent analysis of the pancreatic adenocarcinoma progression model using a pancreatic intraepithelial neoplasia tissue microarray. Mod Pathol 2003; 16: 902-12. ArticlePubMed
- 37. van Heek NT, Meeker AK, Kern SE, et al. Telomere shortening is nearly universal in pancreatic intraepithelial neoplasia. Am J Pathol 2002; 161: 1541-7. ArticlePubMedPMC
- 38. Murphy SJ, Hart SN, Lima JF, et al. Genetic alterations associated with progression from pancreatic intraepithelial neoplasia to invasive pancreatic tumor. Gastroenterology 2013; 145: 1098-109.e1. ArticlePubMedPMC
- 39. Amato E, Molin MD, Mafficini A, et al. Targeted next-generation sequencing of cancer genes dissects the molecular profiles of intraductal papillary neoplasms of the pancreas. J Pathol 2014; 233: 217-27. ArticlePubMedPMCPDF
- 40. Biankin AV, Biankin SA, Kench JG, et al. Aberrant p16(INK4A) and DPC4/Smad4 expression in intraductal papillary mucinous tumours of the pancreas is associated with invasive ductal adenocarcinoma. Gut 2002; 50: 861-8. ArticlePubMedPMC
- 41. Iacobuzio-Donahue CA, Klimstra DS, Adsay NV, et al. Dpc-4 protein is expressed in virtually all human intraductal papillary mucinous neoplasms of the pancreas: comparison with conventional ductal adenocarcinomas. Am J Pathol 2000; 157: 755-61. PubMedPMC
- 42. Satoh K, Shimosegawa T, Moriizumi S, Koizumi M, Toyota T. K-ras mutation and p53 protein accumulation in intraductal mucin-hypersecreting neoplasms of the pancreas. Pancreas 1996; 12: 362-8. ArticlePubMed
- 43. Kawahira H, Kobayashi S, Kaneko K, Asano T, Ochiai T. p53 protein expression in intraductal papillary mucinous tumors (IPMT) of the pancreas as an indicator of tumor malignancy. Hepatogastroen terology 2000; 47: 973-7.
- 44. Chadwick B, Willmore-Payne C, Layfield LJ, Hirschowitz S, Holden J. Histologic, immunohistochemical, and molecular classification of 52 IPMNs of the pancreas. Appl Immunohistochem Mol Morphol 2009; 17: 31-9. ArticlePubMed
- 45. Lubezky N, Ben-Haim M, Marmor S, et al. High-throughput mutation profiling in intraductal papillary mucinous neoplasm (IPMN). J Gastrointest Surg 2011; 15: 503-11. ArticlePubMedPDF
- 46. Dal Molin M, Matthaei H, Wu J, et al. Clinicopathological correlates of activating GNAS mutations in intraductal papillary mucinous neoplasm (IPMN) of the pancreas. Ann Surg Oncol 2013; 20: 3802-8. ArticlePubMedPMCPDF
- 47. Wu J, Jiao Y, Dal Molin M, et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc Natl Acad Sci U S A 2011; 108: 21188-93. ArticlePubMedPMC
- 48. Schönleben F, Qiu W, Ciau NT, et al. PIK3CA mutations in intraductal papillary mucinous neoplasm/carcinoma of the pancreas. Clin Cancer Res 2006; 12: 3851-5. ArticlePubMedPMCPDF
- 49. Sato N, Rosty C, Jansen M, et al. STK11/LKB1 Peutz-Jeghers gene inactivation in intraductal papillary-mucinous neoplasms of the pancreas. Am J Pathol 2001; 159: 2017-22. ArticlePubMedPMC
- 50. Yip-Schneider MT, Wu H, Dumas RP, et al. Vascular endothelial growth factor, a novel and highly accurate pancreatic fluid biomarker for serous pancreatic cysts. J Am Coll Surg 2014; 218: 608-17. ArticlePubMed
- 51. Moore PS, Zamboni G, Brighenti A, et al. Molecular characterization of pancreatic serous microcystic adenomas: evidence for a tumor suppressor gene on chromosome 10q. Am J Pathol 2001; 158: 317-21. PubMedPMC
- 52. Reddy S, Cameron JL, Scudiere J, et al. Surgical management of solid-pseudopapillary neoplasms of the pancreas (Franz or Hamoudi tumors): a large single-institutional series. J Am Coll Surg 2009; 208: 950-7. ArticlePubMedPMC
- 53. Law JK, Ahmed A, Singh VK, et al. A systematic review of solid-pseudopapillary neoplasms: are these rare lesions? Pancreas 2014; 43: 331-7. PubMedPMC
- 54. Abraham SC, Klimstra DS, Wilentz RE, et al. Solid-pseudopapillary tumors of the pancreas are genetically distinct from pancreatic ductal adenocarcinomas and almost always harbor beta-catenin mutations. Am J Pathol 2002; 160: 1361-9. PubMedPMC
- 55. Tanaka Y, Kato K, Notohara K, et al. Frequent beta-catenin mutation and cytoplasmic/nuclear accumulation in pancreatic solid-pseudopapillary neoplasm. Cancer Res 2001; 61: 8401-4. PubMed
- 56. McCall CM, Shi C, Cornish TC, et al. Grading of well-differentiated pancreatic neuroendocrine tumors is improved by the inclusion of both Ki67 proliferative index and mitotic rate. Am J Surg Pathol 2013; 37: 1671-7. ArticlePubMedPMC
- 57. Jiao Y, Shi C, Edil BH, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011; 331: 1199-203. PubMedPMC
- 58. Öberg K. The genetics of neuroendocrine tumors. Semin Oncol 2013; 40: 37-44. ArticlePubMed
- 59. Heaphy CM, de Wilde RF, Jiao Y, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011; 333: 425.PubMedPMC
- 60. de Wilde RF, Heaphy CM, Maitra A, et al. Loss of ATRX or DAXX expression and concomitant acquisition of the alternative lengthening of telomeres phenotype are late events in a small subset of MEN-1 syndrome pancreatic neuroendocrine tumors. Mod Pathol 2012; 25: 1033-9. ArticlePubMedPMCPDF
- 61. Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med 2011; 364: 514-23. ArticlePubMedPMC
- 62. Serrano J, Goebel SU, Peghini PL, Lubensky IA, Gibril F, Jensen RT. Alterations in the p16INK4a/CDKN2A tumor suppressor gene in gastrinomas. J Clin Endocrinol Metab 2000; 85: 4146-56. ArticlePubMed
- 63. Bartsch D, Hahn SA, Danichevski KD, et al. Mutations of the DPC4/Smad4 gene in neuroendocrine pancreatic tumors. Oncogene 1999; 18: 2367-71. ArticlePubMedPDF
- 64. Yachida S, Vakiani E, White CM, et al. Small cell and large cell neuroendocrine carcinomas of the pancreas are genetically similar and distinct from well-differentiated pancreatic neuroendocrine tumors. Am J Surg Pathol 2012; 36: 173-84. ArticlePubMedPMC
- 65. Wood LD, Klimstra DS. Pathology and genetics of pancreatic neoplasms with acinar differentiation. Semin Diagn Pathol 2014; 31: 491-7. ArticlePubMedPMC
- 66. Jiao Y, Yonescu R, Offerhaus GJ, et al. Whole-exome sequencing of pancreatic neoplasms with acinar differentiation. J Pathol 2014; 232: 428-35. ArticlePubMedPMCPDF
- 67. Abraham SC, Wu TT, Hruban RH, et al. Genetic and immunohistochemical analysis of pancreatic acinar cell carcinoma: frequent allelic loss on chromosome 11p and alterations in the APC/beta-catenin pathway. Am J Pathol 2002; 160: 953-62. PubMedPMC
- 68. Chmielecki J, Hutchinson KE, Frampton GM, et al. Comprehensive genomic profiling of pancreatic acinar cell carcinomas identifies recurrent RAF fusions and frequent inactivation of DNA repair genes. Cancer Discov 2014; 4: 1398-405. ArticlePubMedPDF
- 69. Abraham SC, Wu TT, Klimstra DS, et al. Distinctive molecular genetic alterations in sporadic and familial adenomatous polyposis-associated pancreatoblastomas : frequent alterations in the APC/beta-catenin pathway and chromosome 11p. Am J Pathol 2001; 159: 1619-27. PubMedPMC
- 70. Wiley J, Posekany K, Riley R, et al. Cytogenetic and flow cytometric analysis of a pancreatoblastoma. Cancer Genet Cytogenet 1995; 79: 115-8. ArticlePubMed
- 71. Nagashima Y, Misugi K, Tanaka Y, et al. Pancreatoblastoma: a second report on cytogenetic findings. Cancer Genet Cytogenet 1999; 109: 178-9.
- 72. Barenboim-Stapleton L, Yang X, Tsokos M, et al. Pediatric pancreatoblastoma: histopathologic and cytogenetic characterization of tumor and derived cell line. Cancer Genet Cytogenet 2005; 157: 109-17. ArticlePubMed
- 73. Wang CL, Zhao WH, Yu J, Li S. Fluorescence in situ hybridization analysis of pancreatoblastoma. Pancreas 2009; 38: 223-4. ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- c-Myc inhibition and p21 modulation contribute to unsymmetrical bisacridines-induced apoptosis and senescence in pancreatic cancer cells
Agnieszka Kurdyn, Monika Pawłowska, Ewa Paluszkiewicz, Mirosława Cichorek, Ewa Augustin
Pharmacological Reports.2025; 77(1): 182. CrossRef - Evaluation of the Value of Fine Needle Aspiration Cytology and Cell Morphology in Determining the Histogenesis of Pancreatic Lesions With Review of Literature, Overview and Cytological Experience of 25 Years: Original Research
Soudah Bisharah, Abbas Mahmoud
Health Science Reports.2025;[Epub] CrossRef - Research of immunotherapy in pancreatic cancer
Zetian Li, Y.-T. Yu, P.P. Piccaluga, S. Xie
BIO Web of Conferences.2024; 111: 02026. CrossRef - Pancreatoblastoma with a novel fusion gene ofIQSEC1‐RAF1
Hironori Goto, Yuhki Koga, Kenichi Kohashi, Hiroaki Ono, Junkichi Takemoto, Toshiharu Matsuura, Tatsuro Tajiri, Kenji Ihara, Yoshinao Oda, Shouichi Ohga
Pediatric Blood & Cancer.2023;[Epub] CrossRef - Imaging phenotype using 18F-fluorodeoxyglucose positron emission tomography–based radiomics and genetic alterations of pancreatic ductal adenocarcinoma
Chae Hong Lim, Young Seok Cho, Joon Young Choi, Kyung-Han Lee, Jong Kyun Lee, Ji Hye Min, Seung Hyup Hyun
European Journal of Nuclear Medicine and Molecular Imaging.2020; 47(9): 2113. CrossRef - NOSH-aspirin (NBS-1120) inhibits pancreatic cancer cell growth in a xenograft mouse model: Modulation of FoxM1, p53, NF-κB, iNOS, caspase-3 and ROS
Mitali Chattopadhyay, Ravinder Kodela, Gabriela Santiago, Thuy Tien C. Le, Niharika Nath, Khosrow Kashfi
Biochemical Pharmacology.2020; 176: 113857. CrossRef - Targeting Mutant KRAS in Pancreatic Cancer: Futile or Promising?
Friederike Inga Nollmann, Dietrich Alexander Ruess
Biomedicines.2020; 8(8): 281. CrossRef - Ancillary Techniques in Cytologic Specimens Obtained from Solid Lesions of the Pancreas: A Review
Jonas J. Heymann, Momin T. Siddiqui
Acta Cytologica.2020; 64(1-2): 103. CrossRef - DNA polymerase η mutational signatures are found in a variety of different types of cancer
Igor B. Rogozin, Alexander Goncearenco, Artem G. Lada, Subhajyoti De, Vyacheslav Yurchenko, German Nudelman, Anna R. Panchenko, David N. Cooper, Youri I. Pavlov
Cell Cycle.2018; 17(3): 348. CrossRef - Genetics of Pancreatic Neoplasms and Role of Screening
Venkata S. Katabathina, Omid Y. Rikhtehgar, Anil K. Dasyam, Rohan Manickam, Srinivasa R. Prasad
Magnetic Resonance Imaging Clinics of North America.2018; 26(3): 375. CrossRef - Team work and cytopathology molecular diagnosis of solid pancreatic lesions
Carlo Fabbri, Giulia Gibiino, Adele Fornelli, Vincenzo Cennamo, Daniela Grifoni, Michela Visani, Giorgia Acquaviva, Matteo Fassan, Sirio Fiorino, Silvia Giovanelli, Marco Bassi, Stefania Ghersi, Giovanni Tallini, Elio Jovine, Antonio Gasbarrini, Dario de
Digestive Endoscopy.2017; 29(6): 657. CrossRef - Mutational signatures and mutable motifs in cancer genomes
Igor B. Rogozin, Youri I. Pavlov, Alexander Goncearenco, Subhajyoti De, Artem G. Lada, Eugenia Poliakov, Anna R. Panchenko, David N. Cooper
Briefings in Bioinformatics.2017;[Epub] CrossRef - The evolving field of genomic biomarkers to characterize pancreatic cystic neoplasia by EUS-guided FNA
Ferga C. Gleeson, Michael J. Levy
Gastrointestinal Endoscopy.2016; 83(1): 149. CrossRef - MIA PaCa-2 and PANC-1 – pancreas ductal adenocarcinoma cell lines with neuroendocrine differentiation and somatostatin receptors
Rui Gradiz, Henriqueta C. Silva, Lina Carvalho, Maria Filomena Botelho, Anabela Mota-Pinto
Scientific Reports.2016;[Epub] CrossRef - p53 and p16/p19 Loss Promotes Different Pancreatic Tumor Types from PyMT-Expressing Progenitor Cells
Stephanie Azzopardi, Sharon Pang, David S. Klimstra, Yi-Chieh Nancy Du
Neoplasia.2016; 18(10): 610. CrossRef - MicroRNA-506 participates in pancreatic cancer pathogenesis by targeting PIM3
JUNDONG DU, XI ZHENG, SHOUWANG CAI, ZIMAN ZHU, JINGWANG TAN, BIN HU, ZHIQIANG HUANG, HUABO JIAO
Molecular Medicine Reports.2015; 12(4): 5121. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
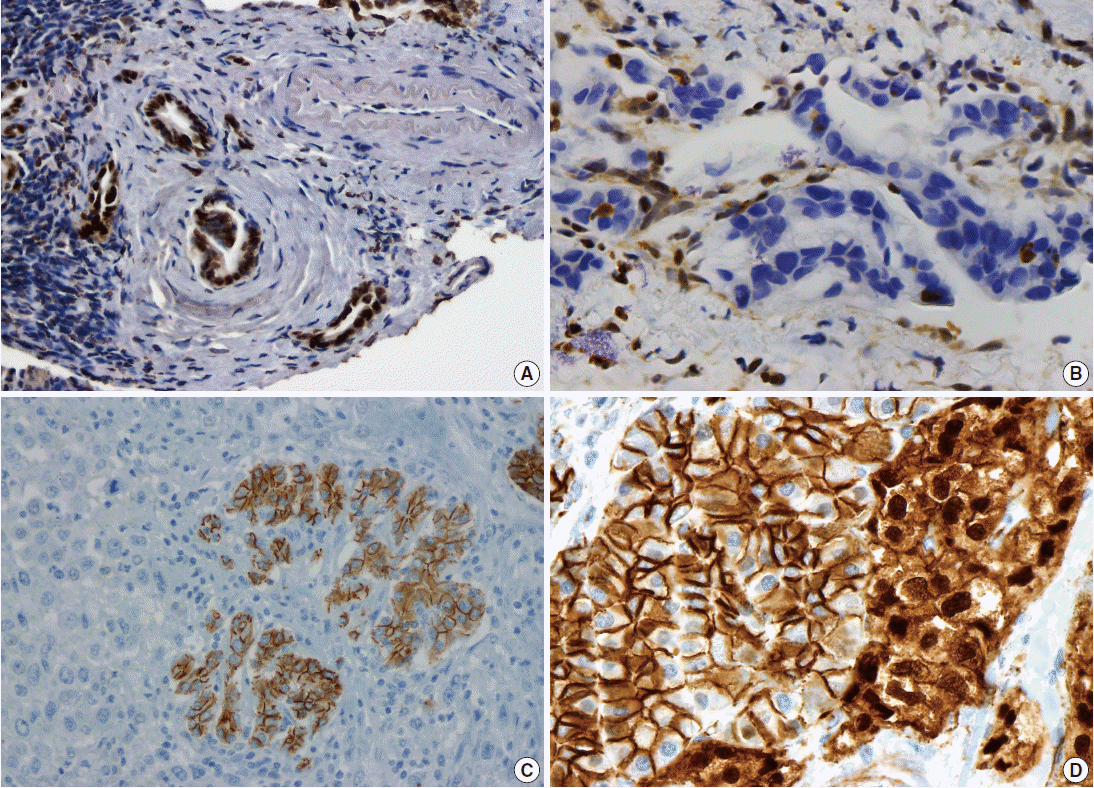

Fig. 1.
| Neoplasm | Gene(s) | Alteration prevalence (%) |
|---|---|---|
| PDAC | KRAS | 95 |
| P16/CDKN2A | 95 | |
| TP53 | 75 | |
| SMAD4/DPC4 | 55 | |
| IPMN | KRAS | 80 |
| RNF43 | 60 | |
| GNAS | 60 | |
| PIK3CA | 10 | |
| P16/CDKN2A | Only in HGD/carcinoma | |
| TP53 | Only in HGD/carcinoma | |
| SMAD4/DPC4 | Only in HGD/carcinoma | |
| MCN | KRAS | 80 |
| RNF43 | 40 | |
| TP53 | Only in HGD/carcinoma | |
| P16/CDKN2A | Only in HGD/carcinoma | |
| SMAD4/DPC4 | Only in HGD/carcinoma | |
| SCA | VHL | 50 |
| SPN | CTNNB1 | 95 |
| PanNET | MEN1 | 45 |
| DAXX/ATRX | 45 | |
| mTOR pathway | 15 | |
| ACC | N umerous genes with nonsynonymous point mutations | 0–30 |
| RAF rearrangements | 25 | |
| PB | CTNNB1 | 55 |
| APC | 10 | |
| 11p loss (gene unknown) | 85 |
| Gene | Syndrome | Neoplasm |
|---|---|---|
| BRCA2 and BRCA1 | Familial breast cancer | PDAC |
| PALB2 (FANCN) | Familial breast cancer | PDAC |
| P16/CDKN2A | Familial atypical multiple mole melanoma syndrome (FAMMM) | PDAC |
| STK11/LKB1 | Peutz-Jeghers syndrome (PJS) | PDAC, IPMN |
| PRSS1, SPINK1 | Hereditary pancreatitis | PDAC |
| hMSH2, hMLH1, hPMS1, hPMS2, hMSH6/GTB | Lynch syndrome/hereditary non-polyposis colorectal cancer (HNPCC) | PDAC (medullary variant) |
| ATM | Ataxia-Telangiectasia | PDAC |
| VHL | von Hippel-Lindau syndrome (VHL) | SCA, PanNET |
| MEN1 | Multiple endocrine neoplasia type 1 (MEN1) | PanNET |
| TSC1, TSC2 | Tuberous sclerosis complex (TSC) | PanNET |
| NF1 | Neurofi omatosis type 1 (NF1) | PanNET |
| Unknown | Beckwith-Wiedemann syndrome (BWS) | PB |
PDAC, pancreatic ductal adenocarcinoma; IPMN, intraductal papillary mucinous neoplasm; HGD, high-grade dysplasia; MCN, mucinous cystic neoplasm; carcinoma, invasive carcinoma; SCA, serous cystadenoma; SPN, solid-pseudopapillary neoplasm; PanNET, well-differentiated pancreatic neuroendocrine tumor; mTOR, mammalian target of rapamycin; ACC, acinar cell carcinoma; PB, pancreatoblastoma.
PDAC, pancreatic ductal adenocarcinoma; IPMN, intraductal papillary mucinous neoplasm; SCA, serous cystadenoma; PanNET, well-differentiated pancreatic neuroendocrine tumor; PB, pancreatoblastoma.

