 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 49(1); 2015 > Article
-
Review
Genomic Landscapes of Pancreatic Neoplasia - Laura D. Wood, Ralph H. Hruban
-
Journal of Pathology and Translational Medicine 2015;49(1):13-22.
DOI: https://doi.org/10.4132/jptm.2014.12.26
Published online: January 15, 2015
The Sol Goldman Pancreatic Cancer Research Center, Johns Hopkins University School of Medicine, Baltimore, MD, USA
- Corresponding Author: Laura D. Wood, M.D. The Sol Goldman Pancreatic Cancer Research Center, Johns Hopkins University School of Medicine, CRB 2 Room 345, 1550 Orleans Street, Baltimore, MD 21231, USA Tel: +1-410-955-3511 Fax: +1-410-614-0671 E-mail: ldwood@jhmi.edu
© 2015 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- Abstract
- PANCREATIC DUCTAL ADENOCARCINOMA AND ITS VARIANTS
- FAMILIAL PANCREATIC CANCER GENES
- PRECURSORS TO PANCREATIC DUCTAL ADENOCARCINOMA: PANCREATIC INTRAEPITHELIAL NEOPLASIAS
- PRECURSORS TO PANCREATIC DUCTAL ADENOCARCINOMA: IPMNS AND MCNS
- OTHER CYSTIC NEOPLASMS
- SOLID-PSEUDOPAPILLARY NEOPLASMS
- PANCREATIC NEUROENDOCRINE TUMORS
- NEOPLASMS WITH ACINAR DIFFERENTIATION
- CONCLUSION
- NOTES
- Acknowledgments
- REFERENCES
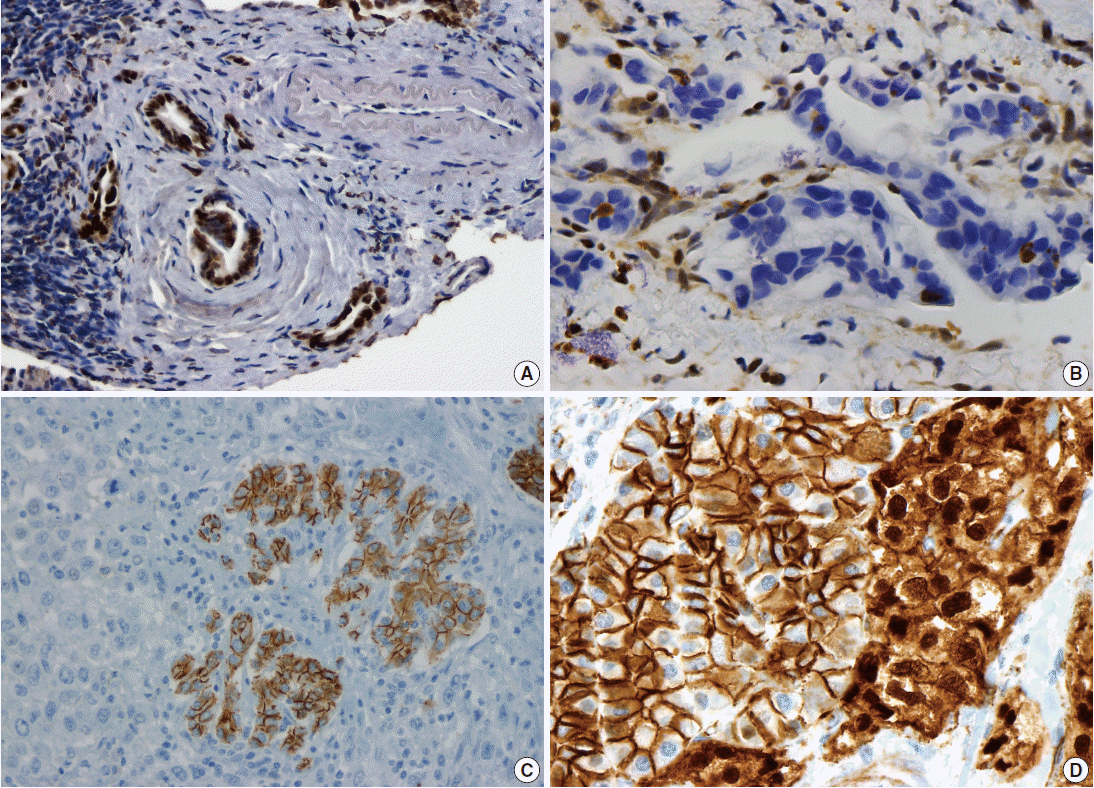
Figure & Data
References
Citations
- c-Myc inhibition and p21 modulation contribute to unsymmetrical bisacridines-induced apoptosis and senescence in pancreatic cancer cells
Agnieszka Kurdyn, Monika Pawłowska, Ewa Paluszkiewicz, Mirosława Cichorek, Ewa Augustin
Pharmacological Reports.2025; 77(1): 182. CrossRef - Evaluation of the Value of Fine Needle Aspiration Cytology and Cell Morphology in Determining the Histogenesis of Pancreatic Lesions With Review of Literature, Overview and Cytological Experience of 25 Years: Original Research
Soudah Bisharah, Abbas Mahmoud
Health Science Reports.2025;[Epub] CrossRef - Research of immunotherapy in pancreatic cancer
Zetian Li, Y.-T. Yu, P.P. Piccaluga, S. Xie
BIO Web of Conferences.2024; 111: 02026. CrossRef - Pancreatoblastoma with a novel fusion gene ofIQSEC1‐RAF1
Hironori Goto, Yuhki Koga, Kenichi Kohashi, Hiroaki Ono, Junkichi Takemoto, Toshiharu Matsuura, Tatsuro Tajiri, Kenji Ihara, Yoshinao Oda, Shouichi Ohga
Pediatric Blood & Cancer.2023;[Epub] CrossRef - Imaging phenotype using 18F-fluorodeoxyglucose positron emission tomography–based radiomics and genetic alterations of pancreatic ductal adenocarcinoma
Chae Hong Lim, Young Seok Cho, Joon Young Choi, Kyung-Han Lee, Jong Kyun Lee, Ji Hye Min, Seung Hyup Hyun
European Journal of Nuclear Medicine and Molecular Imaging.2020; 47(9): 2113. CrossRef - NOSH-aspirin (NBS-1120) inhibits pancreatic cancer cell growth in a xenograft mouse model: Modulation of FoxM1, p53, NF-κB, iNOS, caspase-3 and ROS
Mitali Chattopadhyay, Ravinder Kodela, Gabriela Santiago, Thuy Tien C. Le, Niharika Nath, Khosrow Kashfi
Biochemical Pharmacology.2020; 176: 113857. CrossRef - Targeting Mutant KRAS in Pancreatic Cancer: Futile or Promising?
Friederike Inga Nollmann, Dietrich Alexander Ruess
Biomedicines.2020; 8(8): 281. CrossRef - Ancillary Techniques in Cytologic Specimens Obtained from Solid Lesions of the Pancreas: A Review
Jonas J. Heymann, Momin T. Siddiqui
Acta Cytologica.2020; 64(1-2): 103. CrossRef - DNA polymerase η mutational signatures are found in a variety of different types of cancer
Igor B. Rogozin, Alexander Goncearenco, Artem G. Lada, Subhajyoti De, Vyacheslav Yurchenko, German Nudelman, Anna R. Panchenko, David N. Cooper, Youri I. Pavlov
Cell Cycle.2018; 17(3): 348. CrossRef - Genetics of Pancreatic Neoplasms and Role of Screening
Venkata S. Katabathina, Omid Y. Rikhtehgar, Anil K. Dasyam, Rohan Manickam, Srinivasa R. Prasad
Magnetic Resonance Imaging Clinics of North America.2018; 26(3): 375. CrossRef - Team work and cytopathology molecular diagnosis of solid pancreatic lesions
Carlo Fabbri, Giulia Gibiino, Adele Fornelli, Vincenzo Cennamo, Daniela Grifoni, Michela Visani, Giorgia Acquaviva, Matteo Fassan, Sirio Fiorino, Silvia Giovanelli, Marco Bassi, Stefania Ghersi, Giovanni Tallini, Elio Jovine, Antonio Gasbarrini, Dario de
Digestive Endoscopy.2017; 29(6): 657. CrossRef - Mutational signatures and mutable motifs in cancer genomes
Igor B. Rogozin, Youri I. Pavlov, Alexander Goncearenco, Subhajyoti De, Artem G. Lada, Eugenia Poliakov, Anna R. Panchenko, David N. Cooper
Briefings in Bioinformatics.2017;[Epub] CrossRef - The evolving field of genomic biomarkers to characterize pancreatic cystic neoplasia by EUS-guided FNA
Ferga C. Gleeson, Michael J. Levy
Gastrointestinal Endoscopy.2016; 83(1): 149. CrossRef - MIA PaCa-2 and PANC-1 – pancreas ductal adenocarcinoma cell lines with neuroendocrine differentiation and somatostatin receptors
Rui Gradiz, Henriqueta C. Silva, Lina Carvalho, Maria Filomena Botelho, Anabela Mota-Pinto
Scientific Reports.2016;[Epub] CrossRef - p53 and p16/p19 Loss Promotes Different Pancreatic Tumor Types from PyMT-Expressing Progenitor Cells
Stephanie Azzopardi, Sharon Pang, David S. Klimstra, Yi-Chieh Nancy Du
Neoplasia.2016; 18(10): 610. CrossRef - MicroRNA-506 participates in pancreatic cancer pathogenesis by targeting PIM3
JUNDONG DU, XI ZHENG, SHOUWANG CAI, ZIMAN ZHU, JINGWANG TAN, BIN HU, ZHIQIANG HUANG, HUABO JIAO
Molecular Medicine Reports.2015; 12(4): 5121. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
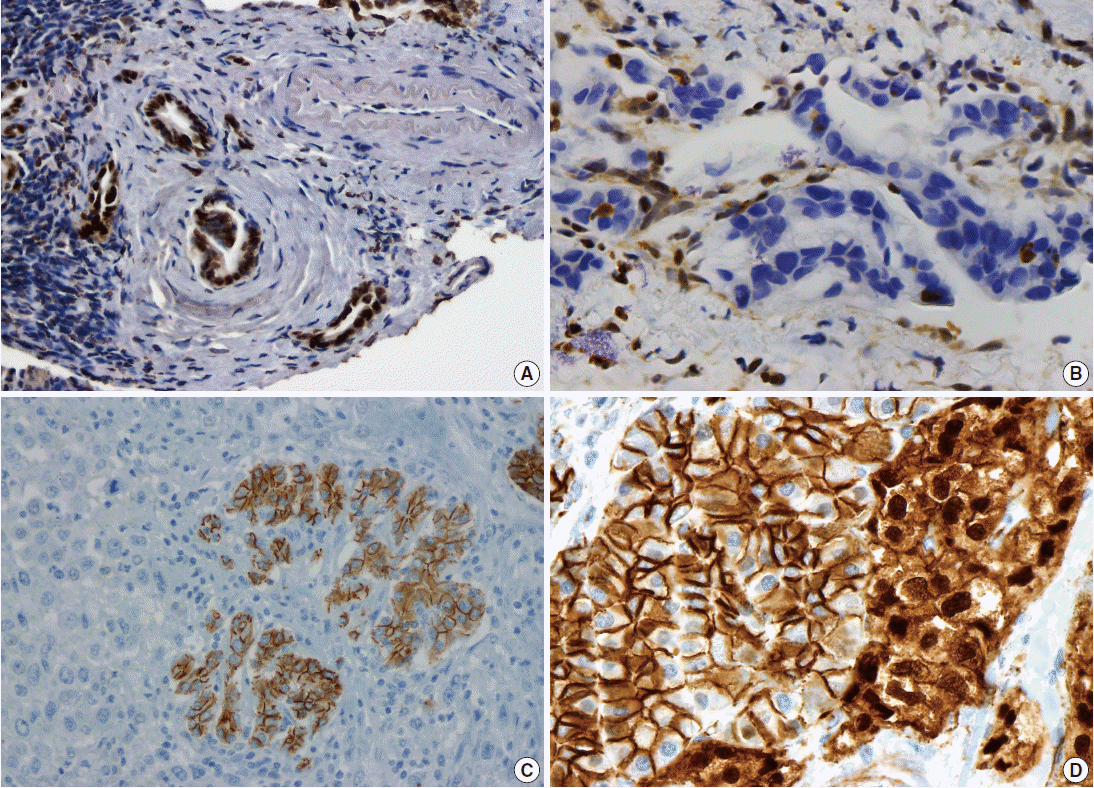

Fig. 1.
| Neoplasm | Gene(s) | Alteration prevalence (%) |
|---|---|---|
| PDAC | KRAS | 95 |
| P16/CDKN2A | 95 | |
| TP53 | 75 | |
| SMAD4/DPC4 | 55 | |
| IPMN | KRAS | 80 |
| RNF43 | 60 | |
| GNAS | 60 | |
| PIK3CA | 10 | |
| P16/CDKN2A | Only in HGD/carcinoma | |
| TP53 | Only in HGD/carcinoma | |
| SMAD4/DPC4 | Only in HGD/carcinoma | |
| MCN | KRAS | 80 |
| RNF43 | 40 | |
| TP53 | Only in HGD/carcinoma | |
| P16/CDKN2A | Only in HGD/carcinoma | |
| SMAD4/DPC4 | Only in HGD/carcinoma | |
| SCA | VHL | 50 |
| SPN | CTNNB1 | 95 |
| PanNET | MEN1 | 45 |
| DAXX/ATRX | 45 | |
| mTOR pathway | 15 | |
| ACC | N umerous genes with nonsynonymous point mutations | 0–30 |
| RAF rearrangements | 25 | |
| PB | CTNNB1 | 55 |
| APC | 10 | |
| 11p loss (gene unknown) | 85 |
| Gene | Syndrome | Neoplasm |
|---|---|---|
| BRCA2 and BRCA1 | Familial breast cancer | PDAC |
| PALB2 (FANCN) | Familial breast cancer | PDAC |
| P16/CDKN2A | Familial atypical multiple mole melanoma syndrome (FAMMM) | PDAC |
| STK11/LKB1 | Peutz-Jeghers syndrome (PJS) | PDAC, IPMN |
| PRSS1, SPINK1 | Hereditary pancreatitis | PDAC |
| hMSH2, hMLH1, hPMS1, hPMS2, hMSH6/GTB | Lynch syndrome/hereditary non-polyposis colorectal cancer (HNPCC) | PDAC (medullary variant) |
| ATM | Ataxia-Telangiectasia | PDAC |
| VHL | von Hippel-Lindau syndrome (VHL) | SCA, PanNET |
| MEN1 | Multiple endocrine neoplasia type 1 (MEN1) | PanNET |
| TSC1, TSC2 | Tuberous sclerosis complex (TSC) | PanNET |
| NF1 | Neurofi omatosis type 1 (NF1) | PanNET |
| Unknown | Beckwith-Wiedemann syndrome (BWS) | PB |
PDAC, pancreatic ductal adenocarcinoma; IPMN, intraductal papillary mucinous neoplasm; HGD, high-grade dysplasia; MCN, mucinous cystic neoplasm; carcinoma, invasive carcinoma; SCA, serous cystadenoma; SPN, solid-pseudopapillary neoplasm; PanNET, well-differentiated pancreatic neuroendocrine tumor; mTOR, mammalian target of rapamycin; ACC, acinar cell carcinoma; PB, pancreatoblastoma.
PDAC, pancreatic ductal adenocarcinoma; IPMN, intraductal papillary mucinous neoplasm; SCA, serous cystadenoma; PanNET, well-differentiated pancreatic neuroendocrine tumor; PB, pancreatoblastoma.

