 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 54(1); 2020 > Article
-
Review
Tumor immune response and immunotherapy in gastric cancer -
Yoonjin Kwak1,2
 , An Na Seo3, Hee Eun Lee4, Hye Seung Lee2,5
, An Na Seo3, Hee Eun Lee4, Hye Seung Lee2,5 -
Journal of Pathology and Translational Medicine 2020;54(1):20-33.
DOI: https://doi.org/10.4132/jptm.2019.10.08
Published online: November 1, 2019
1Department of Pathology, Seoul National University Hospital, Seoul, Korea
2Department of Pathology, Seoul National University College of Medicine, Seoul, Korea
3Department of Pathology, School of Medicine, Kyungpook National University, Kyungpook National University Chilgok Hospital, Daegu, Korea
4Division of Anatomic Pathology, Mayo Clinic, Rochester, MN, USA
5Department of Pathology, Seoul National University Bundang Hospital, Seongnam, Korea
- Corresponding Author: Hye Seung Lee, MD, PhD, Department of Pathology, Seoul National University Bundang Hospital, Seoul National University College of Medicine, 82 Gumi-ro 173beon-gil, Bundang-gu, Seongnam 13620, Korea Tel: +82-31-787-7714, Fax: +82-31-787-4012, E-mail: hye2@snu.ac.kr
© The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- Remarkable developments in immuno-oncology have changed the landscape of gastric cancer (GC) treatment. Because immunotherapy intervenes with tumor immune response rather than directly targeting tumor cells, it is important to develop a greater understanding of tumor immunity. This review paper summarizes the tumor immune reaction and immune escape mechanisms while focusing on the role of T cells and their co-inhibitory signals, such as the immune checkpoint molecules programmed death-1 and programmed deathligand 1 (PD-L1). This paper also describes past clinical trials of immunotherapy for patients with GC and details their clinical implications. Strong predictive markers are essential to improve response to immunotherapy. Microsatellite instability, Epstein-Barr virus, PD-L1 expression, and tumor mutational burden are now regarded as potent predictive markers for immunotherapy in patients with GC. Novel immunotherapy and combination therapy targeting new immune checkpoint molecules such as lymphocyte-activation gene 3, T cell immunoglobulin, and mucin domain containing-3, and indoleamine 2,3-dioxygenase have been suggested, and trials are ongoing to evaluate their safety and efficacy. Immunotherapy is an important treatment option for patients with GC and has great potential for improving patient outcome, and further research in immuno-oncology should be carried out.
- Tumor immune response
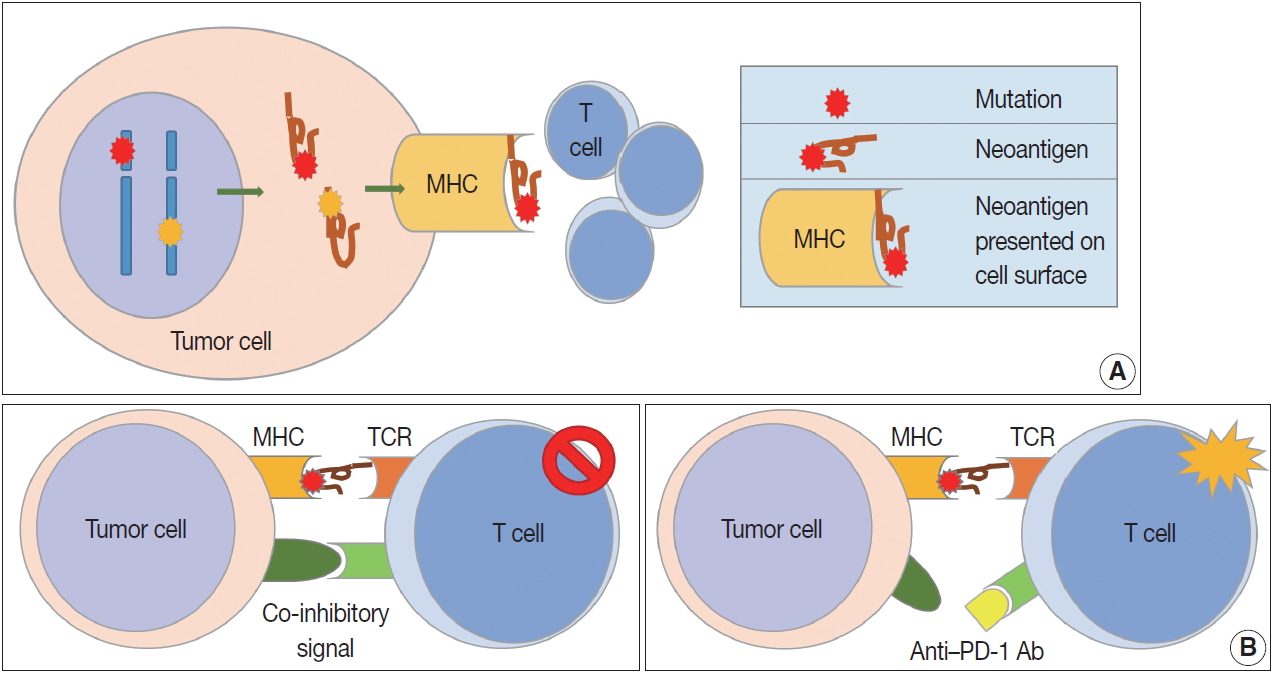
- While the immune response to tumor cells is mainly caused by CD8-positive cytotoxic T cells, it is a multistep process (socalled cancer-immunity cycle) [2,3], including (1) tumor antigen production by tumor cells and processing of released tumor antigens by antigen presenting cells (APCs) such as dendritic cells (DCs), (2) tumor antigen presentation on the surface of APCs through major histocompatibility complex (MHC) class I and MHC class II substances for recognition by T cells, (3) priming and activation of T cells in lymph nodes after recognition of tumor antigens, (4) migration of activated T cells to tumor through blood vessels, (5) infiltration of T cells into and around the tumor, (6) T cell recognition of antigen presenting tumor cells by binding between MHC class I substances on tumor cells and T cell receptors (TCRs) on the surface of T cells, and (7) killing of tumor cells recognized by T cells. The tumor cells destroyed by T cells release additional tumor antigens, increasing immune response. Some receptors and their ligands have been reported to promote or inhibit each step.
- Because patients with tumors have enhanced immune reactions, many researchers have tried to identify tumor antigens that are expressed in tumor cells [4]. Proteins made from gene mutations can be recognized as tumor antigens. As normal cells become tumor cells, mutations are observed in various oncogenes and tumor suppressor genes (Fig. 1A). Furthermore, mutations can easily occur during the gene replication process of cell division due to the genetic instability of tumor cells [5]. Many proteins made from mutated genes are recognized as tumor antigens by immune cells regardless of their function. Neoantigens produced by genetic mutations can induce strong tumor immune responses [6]. Viral proteins from oncogenic viruses, such as human papillomaviruses and Epstein-Barr virus (EBV), are also tumor antigens. In patients infected with the virus, immune cells recognize proteins produced by the virus as antigens, resulting in an immune response to remove the virus-infected cells. Thus, proteins produced from viral genes in tumor cells are important tumor antigens [7]. In addition, proteins that are not expressed in normal cells can be overexpressed in tumor cells. These proteins could be recognized by the immune system as non-self [8]. Some glycoproteins and glycolipids expressed on the cellular surface are overexpressed or modified in tumor cells, which can be recognized as tumor antigens. Oncofetal antigens, such as carcinoembryonic antigen, can also cause immune responses with weak intensity and may be used as diagnostic markers.
- Cell-mediated immunity is the main response to tumors, and the inhibitory effect of cytotoxic T cells on tumor cells is well known [9]. Cytotoxic T cells express CD8 and CD3, and have TCRs that recognize tumor antigens presented by MHC class I molecules [10]. T cell proliferation and activation in tumor tissues requires stimulation, usually by two stimuli (Fig. 1B). The first stimulus is a tumor antigen presented on MHC molecules. As described above, neoantigens from genetically mutated or viral genes are major tumor antigens that cause tumor antigen-specific T cell responses. Second, co-stimulatory molecules affect T cell proliferation and survival. T cells have co-stimulatory receptors that combine with expressed co-stimulatory ligands on the surface of tumor cells to promote T cell activation. CD28 and CD27 expression is observed in T cells, and CD134 and CD137 are mainly expressed in antigen-activated T cells [11,12]. Helper T cells are positive for CD4 and CD3 expression. The role of helper T cells in tumor immunity has yet to be fully elucidated, but their main role is to aid humoral immunity via B cell antibody secretion and to enhance the function of cytotoxic T cells [13]. Helper T cells are reported to have cytotoxic functions in some tumors expressing MHC class II. Regulatory T cells (Tregs) express CD4, FOXP3, and CD25 and regulate the immune response by suppressing the immune response to self-proteins and the tumor immune response [14]. Tregs inhibit the tumor immune response by producing high-affinity interleukin-2 (IL-2) receptor, cytotoxic T lymphocyte–associated protein 4 (CTLA-4), IL-10, and immunosuppressive cytokines such as transforming growth factor β (TGF-β).
- Mechanism of immune evasion in solid tumors
- The immune surveillance system attempts to eliminate tumor cells in the early stages of cancer. However, tumor cells can inhibit the immune system or cause tumor cells themselves to avoid immune responses, a process called immunoediting [15]. Tumor cells can adapt to avoid the immune response by various mechanisms. They can then reach an equilibrium state and escape from the tumor immune response [16].
- In contrast to the co-stimulatory receptors described above, tumor cells produce several co-inhibitory receptors, including PD-1, CTLA-4, T cell immunoglobulin, and mucin domain containing-3 (TIM-3), lymphocyte-activation gene 3 (LAG-3), and indoleamine 2,3-dioxygenase (IDO). These receptors inhibit T cell activation by binding to their tumor cell ligands [11,12]. Tumor-infiltrating lymphocytes (TILS) release interferon γ (IFN-γ) and induce expression of PD-L1 (also known as B7-H1 or CD274) in surrounding tumor cells, stromal cells, and blood cells [17]. Overexpressed PD-L1 activates and binds to PD-1 co-inhibitory receptors on CD8-positive T cells, rendering them inactive. In addition to PD-L1, PD-L2 (also known as B7-DC or CD273) also binds to PD-1 with a weaker binding strength than PD-L1. CTLA-4 is overexpressed early in CD4-positive and CD8-positive T cells. CTLA-4 binds to CD80 (B7-1) and CD86 (B7-2) on APCs, preventing them from binding to the co-stimulatory receptor CD28 and inhibiting TCRs by intracellular PP2A and SHP-2 signaling pathways [18]. CTLA-4 is continuously expressed in Tregs and supports the immunosuppressive function of Tregs [19]. LAG-3 expression is increased in activated T cells and natural killer cells, and its ligands are MHC class II, LSECtin, and galectin-3 [20,21]. LAG-3 inhibits T cell proliferation and cytokine production. TIM-3 binds to its ligands, including galectin-9, high mobility group box 1 (HMGB1), and carcinoembryonic antigen cell adhesion molecule 1, and plays a role in immune evasion of tumor cells by inactivating T cells [22].
- MHC molecules are observed on the surface of all nucleated cells, and MHC class I molecules consist of a heavy chain (HLA-A, -B, and -C) and β-2-microglobulin (β2M) [23]. MHC class I molecules play a major role in the cell-mediated immunity caused by cytotoxic T cells. Expression of MHC class I molecules is heterogeneously observed in patients with cancer. Downregulation or loss of MHC class I molecule expression has been reported and is regarded as the main mechanism of avoiding immune responses [24]. β2M gene mutation and alterations in HLA gene transcription and translation are thought to be major causes of decreased MHC class I expression [25].
- Tumor cells produce and secrete immunosuppressive factors that inhibit the function of immune cells, such as IL-10, galectins, tumor necrosis factor, TGF-β, prostaglandin E2, and vascular endothelial growth factor [26]. They not only inhibit the function of immune cells but also interfere with their differentiation and maturation. In addition, tumor cells can evade the immune response by modifying tumor antigens [26]. As a tumor grows, tumor cells with immunogenic tumor antigens are removed by the immune response. The immune response can no longer remove tumor cells that lack tumor antigens.
- Prognostic significance of tumor-infiltrating lymphocytes in gastric cancer
- In recent years, TILs have been studied for their role as prognostic markers and potential therapeutic targets. Neoantigens presented on cancer cells can recruit TILs and trigger an immune reaction. CD8-positive cytotoxic T cells play a role as the effector cytotoxic T cells involved in direct killing of tumor cells [5]. Therefore, they are regarded as the anti-tumorigenic T cell population. Many previous studies reported that higher TIL density was associated with favorable prognosis in patients with cancer, including GC. Furthermore, previous studies consistently demonstrated that higher TIL densities, such as CD3- or CD8-positive cytotoxic T cells, were associated with better outcome in patients with GC [27,28]. These studies investigated TIL density by immunohistochemistry (IHC) of CD3, CD4, CD8, and other markers (Table 1) [29-42]. An image analyzing software was used to quantify TIL density and ensure constant analysis, or TIL density was counted manually. Most studies dichotomized TIL density into a low and high group for statistical analysis. However, the detailed methods and cut-offs are diverse and not yet standardized. For example, analysis areas were selected in various ways and cut-off values for TIL density had a diverse range. TILs were counted in one representative area, two to six representative areas, or in both the center and at the invasive border. Some studies defined cutoffs as a median value, but others defined cutoffs as a mean, 25th percentile, or 60th percentile value calculated in their own cohort. Thus, the cutoff number for CD8-positive cytotoxic T cells ranged from 21.6/mm2 to 946.22/mm2. Although higher TIL density is repeatedly reported as a favorable prognostic biomarker, diagnostic methods are not standardized and there is no consensus regarding the cutoff for high TIL density. Therefore, further study and consensus are needed to clarify the diagnostic reliability and practical usefulness of TIL densities in patients with GC.
TUMOR IMMUNE MICROENVIRONMENT: GENERAL CONCEPTS AND PROGNOSTIC SIGNIFICANCE
- GC is the fourth most common cancer in the world and the second-leading cause of cancer-associated death [43]. For localized GC, the only curative treatment modality is radical surgery with or without perioperative chemotherapy [44]. For unresectable or metastatic GC, systemic chemotherapy has been the mainstay of palliation. Over the last decade, two new agents, monoclonal antibodies targeting human epidermal growth factor receptor 2 (HER2; trastuzumab) and vascular endothelial growth factor receptor 2 (VEGFR-2; ramucirumab), have been approved by the FDA to treat GC in the palliative setting [44]. Briefly, first-line therapy includes a combination of platinum and fluoropyrimidine (5-fluorouracil or capecitabine) with trastuzumab added in HER2-positive tumors [44]. In the second-line setting, therapeutic options include docetaxel, paclitaxel, or irinotecan monotherapy or ramucirumab alone or in combination with paclitaxel [44]. In fact, nearly all patients with advanced GC experience disease progression following treatment [45]. There is currently no standard third-line therapy but options include apatinib, a small molecule multitargeted tyrosine kinase inhibitor with activity against VEGFR-2, and regorafenib, a multikinase inhibitor [44]. More recently, immune checkpoint inhibitors have emerged as among the most advanced therapeutic options available for patients with advanced GC [46].
- Pembrolizumab is a selective, humanized, high-affinity IgG4 kappa monoclonal antibody that binds to PD-1, blocking its interaction with PD-L1 and 2 [45,47]. The FDA approved pembrolizumab in May 2017 for patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors that progressed even after prior treatment and who have no other optimal treatment choices [48]. The approval was based on promising results of five different clinical trials; the overall/objective response rate (ORR), defined as proportion of patients with a complete or partial response, was 39.6% among 149 patients with various tumor types (95% confidence interval [CI], 31.7% to 47.9%). A complete response was seen in 7% of patients [48]. The duration of response ranged from 1.6+ to 22.7+ months (+indicates no progressive disease at last patient assessment) [48]. The phase 1b KEYNOTE-012 trial had ORR of 22% among 36 patients with PD-L1 positive recurrent or metastatic gastric or gastroesophageal (G/GEJ) adenocarcinomas, and all responses were partial [49]. The phase 2 KEYNOTE-059 trial enrolled 259 patients with locally advanced or metastatic G/GEJ adenocarcinoma to further assess the safety and efficacy of pembrolizumab [50]. The ORR was 11.6% (95% CI, 8.0% to 16.1%), with complete response in 2.3% of cases (95% CI, 0.9% to 5.0%), and the response duration ranged from 1.6+ to 17.3+ months. Among 55% of the tumors with PD-L1 expression based on a combined positive score (CPS)≥1 and that were either microsatellite stable or with undetermined microsatellite instability, the ORR was 13.3% (95% CI, 8.2 to 20.0), with complete response in 1.4% of cases. The duration of response was relatively long, ranging from 2.8+ to 19.4+ months. Based on these results, the FDA granted approval with acceleration for pembrolizumab to treat patients with recurrent, locally advanced or metastatic G/GEJ adenocarcinoma with disease progression on or after two or more standard systemic therapies and if tumors express PD-L1, as determined by an FDA-approved test (PD-L1 IHC 22C3 pharmDx test, Dako, Agilent, Santa Clara, CA, USA) [47]. In the phase 3 KEYNOTE-061 trial, pembrolizumab did not demonstrate significant improvement in overall survival (OS) compared to paclitaxel as second-line therapy for advanced G/GEJ cancer with PD-L1 CPS≥1 [51]. However, pembrolizumab had a better safety profile than paclitaxel. The phase 3 KEYNOTE-062 trial enrolled 763 patients with PD-L1–positive, HER2-negative, advanced G/GEJ cancer; pembrolizumab used as a first-line therapy resulted in noninferior OS compared with standard chemotherapy [52]. Additionally, pembrolizumab showed significant improvement in OS among patients that had tumors with PD-L1 CPS ≥10.
- Nivolumab, a human IgG4 monoclonal antibody that inhibits PD-1, has been approved for monotherapy and combination therapy in metastatic melanoma, non-small cell lung cancer, and renal cell carcinoma [46]. The phase 1/2 CheckMate-032 trial showed ORR of 12% in the nivolumab alone group in patients with chemotherapy-refractory G/GEJ cancer [53]. The phase 3 ATTRACTION-2 trial demonstrated significantly longer OS in the nivolumab alone group vs placebo (5.26 months vs. 4.14 months) in patients with advanced G/GEJ cancer refractory to at least two previous chemotherapies [45]. As a consequence, nivolumab was approved to treat advanced GC as a third-line therapy in Japan [46]. In addition, interim results of the phase 2 ATTRACTION-4 trial showed that nivolumab combined with standard chemotherapy showed promising efficacy as a first-line therapy for unresectable advanced or recurrent HER2-negative G/GEJ cancer [54]. The ATTRACTION-4 trial has proceeded to part 2 (phase 3) to compare nivolumab plus standard chemotherapy versus placebo plus standard chemotherapy. Another phase 3 trial (CheckMate-649) is ongoing to evaluate the efficacy and safety of nivolumab combined with standard chemotherapy or ipilimumab (anti-CTLA-4) as a first-line therapy in patients with advanced G/GEJ cancer [55].
- There are many other ongoing clinical trials to evaluate the safety and efficacy of a variety of immune checkpoint inhibitors in patients with advanced G/GEJ cancer. These include tremelimumab (anti-CTLA-4) [56], avelumab (anti-PD-L1) [57,58], durvalumab (anti-PD-L1) [56,59], and relatlimab (anti-LAG3) [60]. Most of the trials are currently ongoing. A completed trial (JAVELIN Gastric 300) showed that treatment of patients with G/GEJ cancer with single-agent avelumab in the third-line setting did not demonstrate improved OS or progression-free survival (PFS) compared with standard chemotherapy [57]. The completed and ongoing immunotherapy clinical trials in locally advanced, recurrent, or metastatic G/GEJ cancers are summarized in Table 2.
CURRENT STATUS OF IMMUNOTHERAPY FOR GASTRIC CANCER
- IHC of PD-L1
- PD-1 was first discovered in 1992 by Honjo and colleagues [61]. PD-1 is mainly expressed on activated cytotoxic T cells and other immune cells [51,62]. Within the tumor microenvironment, PD-L1 and PD-L2 can upregulate their expression in cancer cells [62]. In several previous studies and currently published genomic profiles, PD-L1 expression has been observed in more than 40% of GC; it is particularly specific for EBV-positive and MSI-H subtypes [37,63-65].
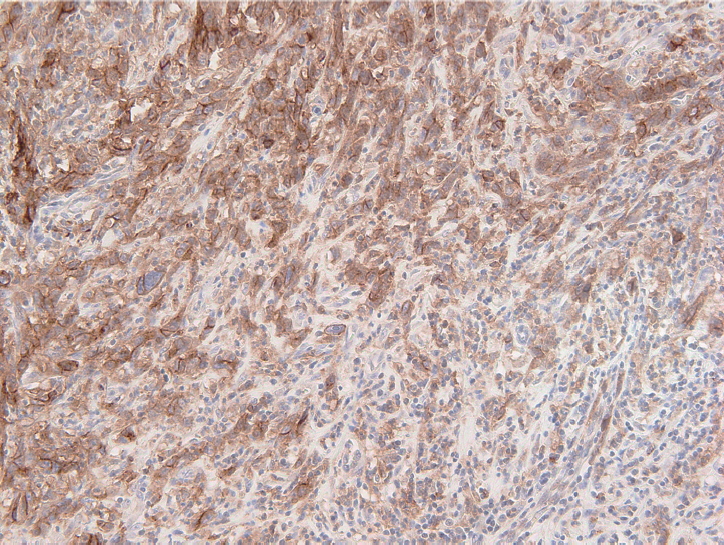
- The PD-L1 IHC 22C3 pharmDx assay on the Autostainer Link 48 platform is the FDA-approved companion diagnostic assay to help identify recurrent or metastatic G/GEJ adenocarcinoma in patients who could be treated with pembrolizumab [49,50]. PD-L1 expression assessed by this assay can be quantified by the CPS method (Fig. 2), which is the number of PD-L1 staining cells (tumor cells, lymphocytes, and macrophages) divided by the total number of viable tumor cells, multiplied by 100. If the result of the calculation exceeds 100, the maximum score is regarded as CPS 100 [66]. A tumor with CPS≥1 score is considered positive for PD-L1 expression. For adequate evaluation, at least 100 viable tumor cells are needed in a stained slide [66]. At 20× magnification, partial or complete membrane staining of viable tumor cells and membrane and/or cytoplasmic staining of mononuclear inflammatory cells within tumor and peri-tumoral areas were evaluated, respectively [66]. Another interpretation of PD-L1 expression is the tumor proportion score (TPS), which is evaluation of membrane staining of PD-L1 expression on tumor cells. The Asian ATTRACTION-2 study, which determined the efficacy of nivolumab in advanced G/GEJ adenocarcinoma, retrospectively evaluated PD-L1 expression by PD-L1 IHC 28-8 pharmDx assay (Dako, Carpinteria, CA, USA); PD-L1 positivity was defined as TPS ≥1 [46]. Of note, the ATTRACTION-2 study showed that nivolumab had significant benefit in all patients, regardless of PD-L1 expression [46]. The PD-L1 IHC 28-8 pharmDx assay gained FDA approval as a complementary diagnostic test for nivolumab as second line treatment for advanced non-small cell lung cancer (NSCLC) [67].
- Microsatellite instability
- MSI-H GC was proposed as a distinct subgroup of GCs by two large scale molecular studies, The Cancer Genome Atlas (TCGA) and the Asian Cancer Research Group classification [65,68,69]. The incidence of MSI-H GCs varies between countries and ethnicity, ranging from 5.6 to 22.7% [65,68,70-73]. MSI-H GCs are associated with antrum location, female gender, relatively older age, earlier stage, and Lauren intestinal type [72]. The presence of dMMR results in tumor cells accumulating frequent genetic mutations. With high mutational burden, tumor cells produce several neo-antigens that trigger T cell activation and recruitment. As the tumor immune reaction increases, expression of checkpoint molecules in tumor cells and immune cells is upregulated [69]. MSI-H GCs are significantly related to both high TILs and high expression of PD-L1 [74,75]. This has been confirmed by several previous genomic profiling studies [76]. On the basis of these hypotheses, recent studies have suggested that MSI-H status may be used as a biomarker for treatment selection with immune-checkpoint inhibitors [50,69]. In Korean patients with metastatic and/or recurrent GC, almost all patients with MSI-H GCs achieve a dramatic response with pembrolizumab alone [75].
- Because accurate and reliable evaluation of MSI status is important for treatment decisions, more sensitive, more precise, and faster techniques are required [69]. Three representative methods are currently used to detect GCs with dMMR: polymerase chain reaction (PCR), IHC, and next-generation sequencing (NGS). First, PCR amplification with specific primers for microsatellite repeats is a conventional method using capillary gel electrophoresis [77]. By comparing the allelic position of a microsatellite locus in tumor tissue and corresponding normal tissue, MSI can be determined by peak patterns with a shift [69,77]. The National Cancer Institute recommends the Bethesda Panel, which is composed of five microsatellite markers specific for two mononucleotide repeats (BAT25 and BAT26) and three dinucleotide repeats (D5S346, D2S123, and D17S250) [69,77,78]. Another MSI test is composed of a pentaplex panel of quasimonomorphic mononucleotide markers (NR-27, NR-21, NR-24, BAT-25, and BAT-26) [77,79]. MSI-H is designated when a size shift is identified in at least two of five microsatellites loci [77]. Second, IHC is assessed by expression or total loss of mismatch repair (MMR) proteins (MLH1, MSH2, PMS2, and MSH6) [77]. Loss of expression in one or more MMR proteins is defined as dMMR. Compared with the PCR-based method, IHC has high sensitivity and specificity (more than 90%, respectively) [77,80], but a combination of both methods increases sensitivity. Although IHC allows the determination of which MMR gene is defective, GCs with MSI-H are mainly caused by epigenetic inactivation of MMR genes (hypermethylation of CpG island methylator pathway and hMLH1 silencing) [65,68,72,73]. Therefore, loss of MLH1 and PMS2 was predominantly found in sporadic GCs (~95.8%), unlike with Lynch syndrome [72]. Interestingly, BRAFV600E mutation has not been found in MSI-H GCs, unlike in MSI-H sporadic colorectal cancer [69,81]. Recently developed NGS-based methods cover a broader range of microsatellite loci. However, NGS has several disadvantages: (1) high investment costs per sample; (2) longer run time; and (3) experiential bioinformatics analysis [69,82].
- Epstein-Barr virus
- EBV-positive GCs constitute a unique subgroup of GCs in the TCGA with several distinct clinicopathologic characteristics, including abundant TILs, male predominance, relatively young age, earlier stage, and favorable prognosis [65,72,73,76]. The incidence of EBV-positive GCs also varies with country and ethnicity, with a range of 2%–20.1% and a worldwide average of nearly 10% [65,72,73,77,83,84]. EBV-positive GCs display CDKN2A (p16INK4A) promoter hyper-methylation, and PD-L1/L2 expression was elevated in genomic profiling, in which IL-12–mediated signaling signatures induced robust presence of immune cells [65,85]. Nearly 50% of EBV-positive GCs showed high expression of PD-L1 [86]. In Korean patients with metastatic and/or recurrent GCs, the patients with EBV-positive GCs achieved dramatic response with pembrolizumab alone [75]. This study suggests that EBV-positive GCs may be good candidates for pembrolizumab monotherapy [75]. Another open-label, multi-arm phase II trial (NCT02951091) is testing the efficacy of nivolumab in EBV-positive GCs as second-line treatment [87]. EBV-positive GCs are commonly accompanied by more extensive infiltration of CD8-positive cytotoxic T cells and IFN-γ, which induce expression of IDO, a potent immune cell inhibitor [76,85]. The IFN-γ driven gene signature, an additional proposed marker of sensitivity to anti–PD-1 treatment, was enriched in EBV-positive GCs [76]. EBV infection demonstrates four latency patterns depending on combinations of latent gene products during the EBV latency cycle: latency Ia, Ib, II, and III. EBV-positive GC demonstrates the latency I pattern, which is limited to EBV-encoded small RNAs (EBERs), BamHI-A rightward transcripts, and Epstein-Barr nuclear antigen 1 [87,88]. The presence of latent membrane protein (LMP) 2A can distinguish latency type Ia or Ib, and LMP2A is expressed in over 50% of EBV-positive GCs [87].
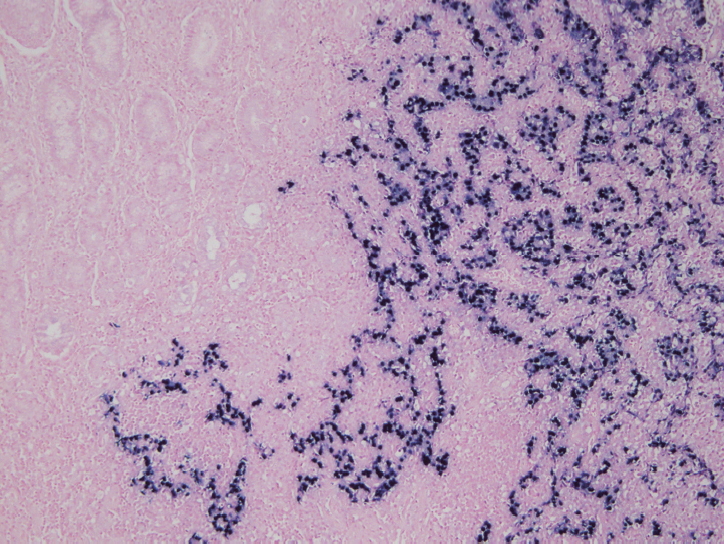
- In situ hybridization (ISH) detection of EBER in tumor cells is considered the gold standard to identify the presence of EBV in tissue samples (Fig. 3) [77,89]. EBER ISH allows examination of paraffin sections from surgical excision and biopsy specimens or on cytology preparations [77]. This method uses light microscopy to demonstrate EBER transcripts concentrated in the nuclei of latently infected cells. Cells are considered EBV positive if brown or blue staining is detected in GC cell nuclei. Commercially available EBER probes are labeled with biotin, digoxigenin, or fluorescein (Ventana Medical Systems, Tucson, AZ, USA; Dako, Glostrup, Denmark; Enzo Diagnostics, Farmingdale, NY, USA; Kreatech Diagnostics, Amsterdam, The Netherlands; Novocastra Laboratories Ltd., Newcastle, UK; Shandon Lipshaw, Pittsburgh, PA, USA; Innogenex, San Ramon, CA, USA) [77,90]. Other methods include PCR or serology comparing EBV nucleic acid positivity or sero-positivity in GC tissues versus non-tumor tissues or in blood of GC patients versus healthy controls [91]. Although IHC is easy and convenient, LMP-1 of representative EBV markers is undetectable in EBV-positive GCs [77].
- Tumor mutational burden
- Tumor mutational burden (TMB), a new predictive biomarker for response to immunotherapy, is a quantitative measure of the total number of somatic nonsynonymous mutations per megabase of genome examined in the DNA of cancer cells [92]. Accumulating data have shown that TMB, or mutational load, is associated with good response to immunotherapy and improved survival [92-96]. Tumors with higher TMB are hypothesized to be more likely to express neoantigens that can be recognized by the immune system in response to immune checkpoint inhibitors [92,97]. The recent CheckMate 227 trial showed that PFS increased in patients with NSCLC treated with nivolumab plus ipilimumab as first-line chemotherapy in tumors with high (≥10 mutations per megabase [Mb]) TMB, regardless of PD-L1 expression [96]. According to this trial, TMB is a promising biomarker for predicting immunotherapy and could lead the way for immuno-oncology entering the era of precision medicine [97]. Clinical trial NCT02915432, which investigated the safety and efficacy of toripalimab in Chinese advanced GC patients, demonstrated that patients with high TMB showed significant treatment response and OS benefit compared with patients with low TMB [98].
- TMB can be measured by whole-exome sequencing (WES) or targeted sequencing panels using NGS [92]. Standardized and stabilized sample processing and methodology are crucial factors that influence the reproducibility of TMB measurements [92]. Initial TMB measurements were assessed by WES using tumor tissue and corresponding non-tumor tissue. Because most WES protocols require a minimum of 150–200 ng of genomic DNA, a limited amount of DNA in small biopsy samples is sometimes problematic [92]. The ability to detect somatic nucleic variants depends on allele frequency within a tumor as well as on sequencing depth [92]. Theoretically, at 50× coverage, if an allele frequency of the specific variant is at least 15%, 95% of single nucleotide variants and short insertions and deletions can consistently be identified [92,99]. Unfortunately, WES has some disadvantages: (1) high cost; (2) extensive analysis; and (3) data management [92]. Gene-targeted sequencing panels using NGS have advantages compared with WES: (1) lower sequencing cost; (2) deeper sequencing despite small amount of DNA; (3) higher sensitivity for mutation detection; and (4) shorter turnaround time [92]. Chalmers et al. [100] demonstrated that comprehensive genomic profiling with the FoundationOne assay targeting ~1.1 Mb of the coding genome can accurately evaluate TMB compared to WES and has high correlation. Some commercial gene panels are now available, such as the Ion Torrent Oncomine Tumor Mutation Load Assay (Thermo Fisher Scientific, Waltham, MA, USA), which targets 1.7 Mb of the genome in 409 key cancer genes.
BIOMARKERS FOR IMMUNOTHERAPY IN GASTRIC CANCER PATIENTS
- While anti–PD-1/PD-L1 therapy has proved successful in GC, many patients do not respond to this treatment or develop resistance. To overcome this limitation of current immunotherapy, a number of ongoing studies are investigating new biomarkers and future strategies for GC treatment. Recently, new immune checkpoints such as LAG-3, TIM-3, and IDO have been suggested as therapeutic targets.
- LAG-3 is a member of the immunoglobulin superfamily that acts as a co-inhibitory receptor expressed on exhausted TILs [101]. It may reduce T cell responses by interacting with MHC class II on antigen presenting cells. A previous study by Woo et al. [102] showed that LAG-3 expression is upregulated on TILs, inhibiting T cell immunity and reducing IFN-γ production within the PD-1 upregulated tumor microenvironment. Co-expression of PD-1 and LAG-3 was demonstrated by in vivo study in a murine cancer model, and LAG-3/PD-1 expression was mostly restricted to TILs. This suggests that combination immunotherapy targeting LAG-3 and PD-1 could magnify tumor-specific responses and avoid non-specific autoimmune responses. Several trials assessing the efficacy of combination therapy of immune checkpoint inhibitors are currently ongoing [103]. The FRACTIONGC trial is investigating whether nivolumab in combination with anti–LAG-3 or anti-IDO inhibitor is more effective than nivolumab in combination with ipilimumab (anti–CTLA-4) in treating patients with advanced GC (NCT 02935634). Another trial starting soon will determine the effectiveness of anti–LAG-3 inhibitor+nivolumab alone or in combination with conventional chemotherapy in participants with G/GEJ adenocarcinoma that recurred or metastasized after prior therapy (NCT03704077).
- TIM-3 is another co-inhibitory receptor expressed on T cells that induces T cell anergy, apoptosis, and exhaustion through interaction with galectin-9 on immune cells [104]. Additionally, TIM-3 can competitively bind to HMGB1 protein, impairing HMGB1-mediated recruitment of nucleic acids into endosomes, an essential step for DNA sensing by the innate immune system [105]. Because of the T cell exhaustion function, TIM-3 has been suggested as an attractive immunotherapy target [106]. A previous study using murine cancer models showed that combined TIM-3/PD-1 blockade is more effective in controlling tumor growth compared to single blockade of TIM-3 or PD-1 [106]. Another in vivo study confirmed that dual blockade of TIM-3 and PD-1 improved the antitumor function of cancer CD8-positive T cells [106]. An ongoing Phase 1 trial is evaluating the safety of anti-TIM-3 inhibitor administered alone or in combination with anti–PD-L1 antibody in participants with advanced or relapsed solid tumors (NCT03099109).
- IDO is a catabolic enzyme produced by Tregs and macrophages to convert tryptophan into kynurenine [107]. IDO-mediated tryptophan deficiency activates naïve T cells and causes them to differentiate into Tregs, causing immune tolerance. In addition, kynurenine converted from tryptophan by IDO also has an immunosuppressive role [108,109]. A study using a melanoma mouse model showed that combinations of CTLA-4 or PD-1/PD-L1 with IDO blockade restored both CD8-positive T cell proliferation and IL-2 production, suggesting the possibility of an IDO inhibitor as a therapeutic option [110]. IDO inhibitors are clinically developing for a variety of cancers, although some trials were stopped due to lack of efficacy [108].
- Because of its central role in tumor immunity, HLA molecules could be biomarkers in future immunotherapy. In a previous study of malignant melanoma, down-regulation of the HLA I molecule was suggested as a potential mechanism of resistance to PD-1 blockade therapy [111]. For sophisticated patient selection, confirmation of HLA I molecule downregulation could become essential. A recent study by Chowell et al. [112] reported that zygosity at HLA loci or certain HLA alleles may influence immunotherapy survival. The authors suggested that greater diversity within the HLA I molecule would result in a larger repertoire of neoantigens for presentation. Through HLA genotype data of cancer patients, homozygosity in at least one HLA class I locus was significantly associated with poor prognosis for immunotherapy. Hence, patient HLA genotype might be utilized as a biomarker for immunotherapy discovery.
BEYOND PD-1/PD-L1: NEW TARGETS OR COMBINATION THERAPY
- Due to important contributions of past research and clinical trials, immunotherapy is now regarded as an important option for treating patients with advanced GC. The mechanism of the tumor immune reaction is complicated; however, understanding of immune responses to cancer cell is crucial for advanced patient selection. MSI and EBV status are regarded as significant prognostic markers in patients with GC. Along with PD-L1 IHC, MSI, and EBV status are now clinically utilized as predictive markers for immunotherapy. Several ongoing studies and clinical trials are validating the efficacy of novel immunotherapy targeting immune checkpoint molecules including LAG-3, TIM-3, and IDO. As demonstrated by previous clinical trials, immunotherapy will greatly improve survival of patients with GC in the future. Thus, research in immuno-oncology should continue, including studies investigating novel biomarkers and clinical trials.
CONCLUSION
Author contributions
Conceptualization: HSL.
Funding acquisition: HSL.
Investigation: YK, ANS, HEL, HSL.
Methodology: YK, ANS, HEL, HSL.
Supervision: HSL.
Writing—original draft: YK, ANS, HEL, HSL.
Writing—review & editing: YK, HSL.
Conflicts of Interest
H.S.L., a contributing editor of the Journal of Pathology and Translational Medicine, was not involved in the editorial evaluation or decision to publish this article. All remaining authors have declared no conflicts of interest.
Funding
This research was supported by a Basic Science Research Program through the National Research Foundation (NRF) funded by the Ministry of Education, Republic of Korea (NRF-2016R1D1A1B03931744).
| Study | Region | No. | Subsets | Outcomes | TMA | Study | Selected area | CD8 cutoff point | CD8 cutoff number (/mm2) |
|---|---|---|---|---|---|---|---|---|---|
| Lee et al. (2008) [29] | Korea | 220 | CD3/CD8/CD45RO | OS | Yes | Consecutive GC | Representative one area | Mean | 435.73 |
| Haas et al. (2009) [30] | Germany | 52 | CD3/CD8/CD20/Foxp3/Granzyme B/M | OS | Yes | Gastric cardia cancer | Six representative areas | Median | 21.6 (epithelial) |
| 212.7 (stromal) | |||||||||
| Shen et al. (2010) [31] | China | 133 | CD4/CD8/Foxp3 | OS | Yes | GC with R0 resection | Average of two centers and two invasive border | Median | 946.22 (intratumoral) |
| 744.40 (peritumoral) | |||||||||
| Kim et al. (2011) [32] | Korea | 180 | CD3/CD4/CD8/Foxp3/Granzyme B | OS/RFS | No | Gastric cardia cancer | Mean of 5 HPFs (center areas) | Median | 60.8/HPF (253.33) |
| Kim et al. (2014) [33] | Korea | 99 | CD8/Foxp3 | OS | Yes | MSI-H advanced GC | Average of 3 representative areas | 60th percentile | 601.5 (median, 542.6) |
| Li e al. (2015) [34] | China | 192 | CD4/CD8 | OS | No | Advanced GC | Representative one slide | 26%-100% staining | Not available |
| Liu et al. (2015) [35] | China | 166 | CD3/CD4/CD8/Foxp3/CD57/M | OS | No | Surgical resection cases | Average of 5 noncontiguous and the densest random areas, in intratumoral and stromal area | Median | 839.69 (intratumoral) |
| 523.90 (stromal) | |||||||||
| Hennequin et al. (2016) [36] | France | 82 | CD8/CD20/Foxp3/Tbet | RFS | No | Consecutive GC (including preop-chemotherapy) | Mean of 3 HPF in core and invasive margin | Median | Not available |
| Kim et al. (2016) [37] | Korea | 243 | CD3/CD4/CD8 | DFS | Yes | Consecutive GC | Representative one core | Median | 375.48 |
| Giampieri et al. (2017) [38] | Italy | 73 | CD3 | OS | No | Metastatic GC with 1st-line chemotherapy | Biopsy or resected specimens | More than 50%-60% stromal area covered by TILs | Not available |
| Kawazoe et al. (2017) [39] | Japan | 383 | CD3/CD4/CD8/Foxp3 | OS | Yes | Advanced GC | Invasive area (two cores) | Median | 384 |
| Koh et al. (2017) [40] | Korea | 392 | CD3/CD4/CD8/Foxp3 | OS | Yes | Stage II and III GC | Tumor center and invasive border | 25th percentile | 130.07 (center) |
| 101.76 (border) | |||||||||
| Pernot et al. (2019) [41] | France | 67 | CD8/Foxp3/CD57 | OS | No | Locally advanced or metastatic GC | Mean of 3 representative HPFs | Median | 31/HPF |
| Kim et al. (2019) [42] | Korea | 297 | CD3/CD8/Foxp3 | OS | Yes | Early GC with submucosal invasion and advanced GC | Tumor center and invasive border | Median | Not available |
| Trials | Target | Phase | Treatment arms | Setting (line) | No. of patients | Results/primary endpoints | |
|---|---|---|---|---|---|---|---|
| Pembralizumab | |||||||
| KEYNOTE-012 [49] | PD-1 | 1b | Pembrolizumab | Any | 39 with positive PD-L1 | ORR (%): 22 (95% CI, 10-39; all PR) | |
| KEYNOTE-059 (cohort 1) [50] | PD-1 | 2 | Pembrolizumab | ≥ 3rd | 259 | ORR (%): 11.6 (95% CI, 8.0–16.1; CR in 2.3) | |
| Median response duration (mo): 8.4 (1.6 +a to 17.3 +) | |||||||
| OS (mo): 5.6; PFS (mo): 2.0 | |||||||
| KEYNOTE-061 [51] | PD-1 | 3 | Pembrolizumab | 2nd | 592 (395 with CPS ≥ 1) | OS (mo): 9.1 vs. 8.3 (HR, 0.82; 95% CI, 0.66-1.03) | |
| PaCIitaxel | PFS (mo): 1.5 vs. 4.1 (HR, 1.27; 95% CI, 1.03-1.57) (both analyses in the subgroup of positive PD-L1) | ||||||
| KEYNOTE-062 [52] | PD-1 | 3 | Pembrolizumab vs | 1st | 763 with CPS ≥ 1 (281 with CPS ≥ 10) | OS (mo): 10.6 vs. 12.5 vs. 11.1 (CPS ≥ 1) | |
| Pembrolizumab + cisplatin + 5-FU or capecitabine | OS (mo): 17.4 vs. 12.3 vs. 10.8 (CPS ≥ 10) | ||||||
| PFS (mo): 2.0 vs. 6.9 vs. 6.4 (CPS ≥ 1) | |||||||
| Placebo + cisplatin + 5-FU or capecitabine | PFS (mo): 2.9 vs. 5.7 vs. 6.1 (CPS ≥ 10) | ||||||
| Nivolumab (± ipilimumab) | |||||||
| CheckMate-032 [53] | PD-1 | 1/2 | Nivolumab 3 mg/kg nivolumab 1 mg/kg + ipilimumab 3 mg/kg | ≥ 2nd | 160 | ORR (%): 12 vs. 24 vs. 8 (independent of PD-L1 status) | |
| CTLA-4 | |||||||
| Nivolumab 3 mg/kg+ipilimumab 1 mg/kg | 12-mo PFS rates (%): 8 vs. 17 vs. 10 | ||||||
| 12-mo OS rates (%): 39 vs. 35 vs. 24 | |||||||
| ONO-4538-12, ATTRACTION-2 [46] | PD-1 | 3 | Nivolumab alone placebo | ≥ 3rd | 493 | OS (mo): 5.26 vs. 4.14 (HR, 0.63; 95% CI, 0.51-0.78) | |
| 12-mo OS rates (%): 26.2 vs. 10.9 | |||||||
| ATTRACTION-4, part 1 [54] | PD-1 | 2 | Nivolumab + oxaliplatin+capecitabine | 1st | 40 | ORR (%): 76.5 vs. 57.1 | |
| Nivdumab+oxaliplatin+S-1 | PFS (mo): 10.6 vs. 9.7 | ||||||
| ATTRACTION-4, part 2 | PD-1 | 3 | Nivolumab + oxaliplatin+S-1 or capecitabine Placebo + oxaliplatin+S-1 or capecitabine | 1st | Approx. 650 | Ongoing | |
| CheckMate-649 [55] | PD-1 | 3 | Nivolumab + ipilimumab | 1st | 870 | Ongoing | |
| CTLA-4 | Nivolumab + oxaliplatin + 5-FU or capecitabine Oxaliplatin + 5-FU or capecitabine | ||||||
| Others | |||||||
| JAVELIN Gastric 100 [58] | PD-L1 | 3 | Avelumab | Maintenance after 1st-line | 499 | Ongoing | |
| BSC after response or stability to oxaliplatinb + fluoropyrimidine | |||||||
| JAVELIN Gastric 300 [57] | PD-L1 | 3 | Avelumab | 3rd | 371 | OS (mo): 4.6 vs. 5.0 (HR, 1.1; 95% CI, 0.9-1.4) | |
| PaCIitaxel or irinotecan | PFS (mo): 1.4 vs. 2.7 (HR, 1.73; 95% CI, 1.4-2.2) | ||||||
| ORR (%): 2.2 vs. 4.3 | |||||||
| NCT02340975 [59] | PD-L1 | 1b/2 | Durvalumab (anti-PD-L1) | ≥ 2nd | 94 (phase 2; as of Sep 13, 2017) | Ongoing | |
| CTLA-4 | Tremelimumab (anti-CTLA-4) | ||||||
| Durvalumab+tremelimumab | |||||||
| NCT01968109 [60] | LAG-3 | 1/2a | Relatlimab (anti-LAG3) vs. Relatlimab + nivolumab | Last | Advanced solid tumors | Ongoing | |
| PD-1 | |||||||
PD-1, programmed death-1; PD-L1, programmed death-ligand 1; ORR, objective response rate; CI, confidence interval; PR, partial response; CR, complete response; OS, overall survival; PFS, progression-free survival; CPS, Combined Positive Score for PD-L1; HR, hazard ratio; CI, confidence interval; 5-FU, 5-fluorouracil; CTLA-4, cytotoxic T lymphocyte–associated protein 4; LAG-3, lymphocyte-activation gene 3.
a+ indicates that patients had no progressive disease at their last assessment.
- 1. Kaufman HL, Atkins MB, Subedi P, et al. The promise of Immunooncology: implications for defining the value of cancer treatment. J Immunother Cancer 2019; 7: 129.ArticlePubMedPMCPDF
- 2. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity 2013; 39: 1-10. ArticlePubMed
- 3. Kim JM, Chen DS. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure). Ann Oncol 2016; 27: 1492-504. ArticlePubMedPDF
- 4. Melief CJ, van Hall T, Arens R, Ossendorp F, van der Burg SH. Therapeutic cancer vaccines. J Clin Invest 2015; 125: 3401-12. ArticlePubMedPMC
- 5. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 2013; 14: 1014-22. ArticlePubMedPMCPDF
- 6. van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat Rev Cancer 2016; 16: 219-33. ArticlePubMedPDF
- 7. Welters MJ, Kenter GG, Piersma SJ, et al. Induction of tumor-specific CD4+ and CD8+ T-cell immunity in cervical cancer patients by a human papillomavirus type 16 E6 and E7 long peptides vaccine. Clin Cancer Res 2008; 14: 178-87. ArticlePubMedPDF
- 8. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer 2014; 14: 135-46. ArticlePubMedPDF
- 9. Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer 2004; 4: 11-22. ArticlePubMedPDF
- 10. Houghton AN, Guevara-Patiño JA. Immune recognition of self in immunity against cancer. J Clin Invest 2004; 114: 468-71. ArticlePubMedPMC
- 11. Schwartz RH. T cell anergy. Annu Rev Immunol 2003; 21: 305-34. ArticlePubMed
- 12. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12: 252-64. ArticlePubMedPMCPDF
- 13. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature 2017; 541: 321-30. ArticlePubMedPDF
- 14. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res 2017; 27: 109-18. ArticlePubMedPDF
- 15. Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol 2011; 29: 235-71. ArticlePubMed
- 16. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol 2004; 22: 329-60. ArticlePubMed
- 17. Taube JM, Anders RA, Young GD, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med 2012; 4: 127ra37.ArticlePubMedPMC
- 18. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol 2016; 39: 98-106. PubMedPMC
- 19. Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory pathways in the B7-CD28 ligand-receptor family. Immunity 2016; 44: 955-72. ArticlePubMedPMC
- 20. Huard B, Prigent P, Tournier M, Bruniquel D, Triebel F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur J Immunol 1995; 25: 2718-21. ArticlePubMed
- 21. Liu B, Wang M, Wang X, et al. Liver sinusoidal endothelial cell lectin inhibits CTL-dependent virus clearance in mouse models of viral hepatitis. J Immunol 2013; 190: 4185-95. ArticlePubMedPDF
- 22. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity 2016; 44: 989-1004. ArticlePubMedPMC
- 23. Parham P, Ohta T. Population biology of antigen presentation by MHC class I molecules. Science 1996; 272: 67-74. ArticlePubMed
- 24. Garrido F, Algarra I. MHC antigens and tumor escape from immune surveillance. Adv Cancer Res 2001; 83: 117-58. ArticlePubMed
- 25. del Campo AB, Kyte JA, Carretero J, et al. Immune escape of cancer cells with beta2-microglobulin loss over the course of metastatic melanoma. Int J Cancer 2014; 134: 102-13. ArticlePubMed
- 26. Kumar V, Abbas A, Aster J. Robbins and Cotran pathologic basis of disease. 9th ed. Philadelphia: Elsevier, 2014.
- 27. Jiang W, Liu K, Guo Q, et al. Tumor-infiltrating immune cells and prognosis in gastric cancer: a systematic review and meta-analysis. Oncotarget 2017; 8: 62312-29. ArticlePubMedPMC
- 28. Zheng X, Song X, Shao Y, et al. Prognostic role of tumor-infiltrating lymphocytes in gastric cancer: a meta-analysis. Oncotarget 2017; 8: 57386-98. ArticlePubMedPMC
- 29. Lee HE, Chae SW, Lee YJ, et al. Prognostic implications of type and density of tumour-infiltrating lymphocytes in gastric cancer. Br J Cancer 2008; 99: 1704-11. ArticlePubMedPMCPDF
- 30. Haas M, Dimmler A, Hohenberger W, Grabenbauer GG, Niedobitek G, Distel LV. Stromal regulatory T-cells are associated with a favourable prognosis in gastric cancer of the cardia. BMC Gastroenterol 2009; 9: 65.ArticlePubMedPMCPDF
- 31. Shen Z, Zhou S, Wang Y, et al. Higher intratumoral infiltrated Foxp3+ Treg numbers and Foxp3+/CD8+ ratio are associated with adverse prognosis in resectable gastric cancer. J Cancer Res Clin Oncol 2010; 136: 1585-95. ArticlePubMedPDF
- 32. Kim HI, Kim H, Cho HW, et al. The ratio of intra-tumoral regulatory T cells (Foxp3+)/helper T cells (CD4+) is a prognostic factor and associated with recurrence pattern in gastric cardia cancer. J Surg Oncol 2011; 104: 728-33. ArticlePubMedPDF
- 33. Kim KJ, Lee KS, Cho HJ, et al. Prognostic implications of tumor-infiltrating FoxP3+ regulatory T cells and CD8+ cytotoxic T cells in microsatellite-unstable gastric cancers. Hum Pathol 2014; 45: 285-93. ArticlePubMed
- 34. Li K, Zhu Z, Luo J, et al. Impact of chemokine receptor CXCR3 on tumor-infiltrating lymphocyte recruitment associated with favorable prognosis in advanced gastric cancer. Int J Clin Exp Pathol 2015; 8: 14725-32. PubMedPMC
- 35. Liu K, Yang K, Wu B, et al. Tumor-infiltrating immune cells are associated with prognosis of gastric cancer. Medicine (Baltimore) 2015; 94: e1631. ArticlePubMedPMC
- 36. Hennequin A, Derangere V, Boidot R, et al. Tumor infiltration by Tbet+ effector T cells and CD20+ B cells is associated with survival in gastric cancer patients. Oncoimmunology 2016; 5: e1054598. ArticlePubMed
- 37. Kim JW, Nam KH, Ahn SH, et al. Prognostic implications of immunosuppressive protein expression in tumors as well as immune cell infiltration within the tumor microenvironment in gastric cancer. Gastric Cancer 2016; 19: 42-52. ArticlePubMedPDF
- 38. Giampieri R, Maccaroni E, Mandolesi A, et al. Mismatch repair deficiency may affect clinical outcome through immune response activation in metastatic gastric cancer patients receiving first-line chemotherapy. Gastric Cancer 2017; 20: 156-63. ArticlePubMedPDF
- 39. Kawazoe A, Kuwata T, Kuboki Y, et al. Clinicopathological features of programmed death ligand 1 expression with tumor-infiltrating lymphocyte, mismatch repair, and Epstein-Barr virus status in a large cohort of gastric cancer patients. Gastric Cancer 2017; 20: 407-15. ArticlePubMedPDF
- 40. Koh J, Ock CY, Kim JW, et al. Clinicopathologic implications of immune classification by PD-L1 expression and CD8-positive tumor-infiltrating lymphocytes in stage II and III gastric cancer patients. Oncotarget 2017; 8: 26356-67. ArticlePubMedPMC
- 41. Pernot S, Terme M, Radosevic-Robin N, et al. Infiltrating and peripheral immune cell analysis in advanced gastric cancer according to the Lauren classification and its prognostic significance. Gastric Cancer 2019 Jul 2 [Epub]. https://doi.org/10.1007/s10120-019-00983-3. ArticlePDF
- 42. Kim JY, Kim WG, Kwon CH, Park DY. Differences in immune contextures among different molecular subtypes of gastric cancer and their prognostic impact. Gastric Cancer 2019; 22: 1164-75. ArticlePubMedPDF
- 43. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015; 65: 87-108. ArticlePubMedPDF
- 44. Charalampakis N, Economopoulou P, Kotsantis I, et al. Medical management of gastric cancer: a 2017 update. Cancer Med 2018; 7: 123-33. ArticlePubMedPDF
- 45. Kiyozumi Y, Iwatsuki M, Yamashita K, Koga Y, Yoshida N, Baba H. Update on targeted therapy and immune therapy for gastric cancer, 2018. J Cancer Metastasis Treat 2018; 4: 31.Article
- 46. Kang YK, Boku N, Satoh T, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017; 390: 2461-71. ArticlePubMed
- 47. Fashoyin-Aje L, Donoghue M, Chen H, et al. FDA approval summary: pembrolizumab for recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma expressing PD-L1. Oncologist 2019; 24: 103-9. ArticlePubMedPDF
- 48. Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA approval summary: pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin Cancer Res 2019; 25: 3753-8. ArticlePubMedPDF
- 49. Muro K, Chung HC, Shankaran V, et al. Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): a multicentre, open-label, phase 1b trial. Lancet Oncol 2016; 17: 717-26. ArticlePubMed
- 50. Fuchs CS, Doi T, Jang RW, et al. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: phase 2 clinical KEYNOTE-059 Trial. JAMA Oncol 2018; 4: e180013. ArticlePubMedPMCPDF
- 51. Shitara K, Özgürog˘lu M, Bang YJ, et al. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): a randomised, open-label, controlled, phase 3 trial. Lancet 2018; 392: 123-33. PubMed
- 52. Tabernero J, Van Custem E, Bang YJ, et al. Pembrolizumab with or without chemotherapy versus chemotherapy for advanced gastric or gastroesophageal junction (G/GEJ) adenocarcinoma: the phase III KEYNOTE-062 study. J Clin Oncol 2019; 37: LBA4007.Article
- 53. Janjigian YY, Bendell J, Calvo E, et al. CheckMate-032 study: efficacy and safety of nivolumab and nivolumab plus ipilimumab in patients with metastatic esophagogastric cancer. J Clin Oncol 2018; 36: 2836-44. ArticlePubMedPMC
- 54. Boku N, Ryu MH, Kato K, et al. Safety and efficacy of nivolumab in combination with S-1/capecitabine plus oxaliplatin in patients with previously untreated, unresectable, advanced, or recurrent gastric/gastroesophageal junction cancer: interim results of a randomized, phase II trial (ATTRACTION-4). Ann Oncol 2019; 30: 250-8. ArticlePubMedPDF
- 55. Janjigian YY, Adenis A, Aucoin JS, et al. Checkmate 649: a randomized, multicenter, open-label, phase 3 study of nivolumab (Nivo) plus ipilimumab (Ipi) versus oxaliplatin plus fluoropyrimidine in patients (Pts) with previously untreated advanced or metastatic gastric (G) or gastroesophageal junction (GEJ) cancer. J Clin Oncol 2017; 35: TPS213.Article
- 56. Kelly RJ, Lee J, Bang YJ, et al. Safety and efficacy of durvalumab in combination with tremelimumab, durvalumab monotherapy, and tremelimumab monotherapy in patients with advanced gastric cancer. J Clin Oncol 2018; 36: 4031.Article
- 57. Bang YJ, Ruiz EY, Van Cutsem E, et al. Phase III, randomised trial of avelumab versus physician's choice of chemotherapy as third-line treatment of patients with advanced gastric or gastro-oesophageal junction cancer: primary analysis of JAVELIN Gastric 300. Ann Oncol 2018; 29: 2052-60. ArticlePubMedPMCPDF
- 58. Moehler M, Ryu MH, Dvorkin M, et al. Maintenance avelumab versus continuation of first-line chemotherapy in gastric cancer: JAVELIN Gastric 100 study design. Future Oncol 2019; 15: 567-77. ArticlePubMed
- 59. A phase 1b/2 study of MEDI4736 with tremelimumab, MEDI4736 or tremelimumab monotherapy in gastric or GEJ adenocarcinoma. Bethesda: ClincalTrials.gov, 2015 [cited 2019 Jul 21]. Available from: https://clinicaltrials.gov/ct2/show/NCT02340975.
- 60. An investigational immuno-therapy study to assess the safety, tolerability and effectiveness of anti-LAG-3 with and without antiPD-1 in the treatment of solid tumors [Internet]. Bethesda: ClincalTrials.gov, 2013 [cited 2019 Jul 21]. Available from: https://clinicaltrials.gov/ct2/show/NCT01968109.
- 61. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 1992; 11: 3887-95. ArticlePubMedPMCPDF
- 62. Ribas A. Tumor immunotherapy directed at PD-1. N Engl J Med 2012; 366: 2517-9. ArticlePubMed
- 63. Qing Y, Li Q, Ren T, et al. Upregulation of PD-L1 and APE1 is associated with tumorigenesis and poor prognosis of gastric cancer. Drug Des Devel Ther 2015; 9: 901-9. PubMedPMC
- 64. Wu C, Zhu Y, Jiang J, Zhao J, Zhang XG, Xu N. Immunohistochemical localization of programmed death-1 ligand-1 (PD-L1) in gastric carcinoma and its clinical significance. Acta Histochem 2006; 108: 19-24. ArticlePubMed
- 65. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014; 513: 202-9. ArticlePubMedPMCPDF
- 66. Interpretation manual: gastric or gastroesophageal junction adenocarcinoma. PD-L1 IHC 22C3 pharmDx interpretation manual: gastric or gastroesophageal juction adenocarcinoma [Internet]. Santa Clara: DAKO Agilent Technologies, 2018 [cited 2019 Jul 21]. Available from: https://www.agilent.com/cs/library/usermanuals/public/29219_pd-l1-ihc-22C3-pharmdx-gastric-interpretation-manual_us.pdf.
- 67. Batenchuk C, Albitar M, Zerba K, et al. A real-world, comparative study of FDA-approved diagnostic assays PD-L1 IHC 28-8 and 22C3 in lung cancer and other malignancies. J Clin Pathol 2018; 71: 1078-83. ArticlePubMedPMC
- 68. Cristescu R, Lee J, Nebozhyn M, et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med 2015; 21: 449-56. ArticlePubMedPDF
- 69. Ratti M, Lampis A, Hahne JC, Passalacqua R, Valeri N. Microsatellite instability in gastric cancer: molecular bases, clinical perspectives, and new treatment approaches. Cell Mol Life Sci 2018; 75: 4151-62. ArticlePubMedPMCPDF
- 70. Polom K, Marano L, Marrelli D, et al. Meta-analysis of microsatellite instability in relation to clinicopathological characteristics and overall survival in gastric cancer. Br J Surg 2018; 105: 159-67. ArticlePubMedPDF
- 71. Kim JY, Shin NR, Kim A, et al. Microsatellite instability status in gastric cancer: a reappraisal of its clinical significance and relationship with mucin phenotypes. Korean J Pathol 2013; 47: 28-35. ArticlePubMedPMCPDF
- 72. Setia N, Agoston AT, Han HS, et al. A protein and mRNA expression-based classification of gastric cancer. Mod Pathol 2016; 29: 772-84. ArticlePubMedPDF
- 73. Ahn S, Lee SJ, Kim Y, et al. High-throughput protein and mRNA expression-based classification of gastric cancers can identify clinically distinct subtypes, concordant with recent molecular classifications. Am J Surg Pathol 2017; 41: 106-15. ArticlePubMed
- 74. Cho J, Chang YH, Heo YJ, et al. Four distinct immune microenvironment subtypes in gastric adenocarcinoma with special reference to microsatellite instability. ESMO Open 2018; 3: e000326.ArticlePubMedPMC
- 75. Kim ST, Cristescu R, Bass AJ, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med 2018; 24: 1449-58. ArticlePubMedPDF
- 76. Lee J, Kim KM. Biomarkers for gastric cancer: molecular classification revisited. Precis Future Med 2017; 1: 59-68. ArticlePDF
- 77. Lee HS, Kim WH, Kwak Y, et al. Molecular testing for gastrointestinal cancer. J Pathol Transl Med 2017; 51: 103-21. ArticlePubMedPMCPDF
- 78. Murphy KM, Zhang S, Geiger T, et al. Comparison of the microsatellite instability analysis system and the Bethesda panel for the determination of microsatellite instability in colorectal cancers. J Mol Diagn 2006; 8: 305-11. ArticlePubMedPMC
- 79. Campanella NC, Berardinelli GN, Scapulatempo-Neto C, et al. Optimization of a pentaplex panel for MSI analysis without control DNA in a Brazilian population: correlation with ancestry markers. Eur J Hum Genet 2014; 22: 875-80. ArticlePubMedPDF
- 80. Bae YS, Kim H, Noh SH, Kim H. Usefulness of immunohistochemistry for microsatellite instability screening in gastric cancer. Gut Liver 2015; 9: 629-35. ArticlePubMedPMCPDF
- 81. Normanno N, Rachiglio AM, Lambiase M, et al. Heterogeneity of KRAS, NRAS, BRAF and PIK3CA mutations in metastatic colorectal cancer and potential effects on therapy in the CAPRI GOIM trial. Ann Oncol 2015; 26: 1710-4. PubMedPDF
- 82. Vanderwalde A, Spetzler D, Xiao N, Gatalica Z, Marshall J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med 2018; 7: 746-56. PubMedPMC
- 83. Lee HS, Chang MS, Yang HK, Lee BL, Kim WH. Epstein-barr viruspositive gastric carcinoma has a distinct protein expression profile in comparison with epstein-barr virus-negative carcinoma. Clin Cancer Res 2004; 10: 1698-705. ArticlePubMedPDF
- 84. Kang BW, Seo AN, Yoon S, et al. Prognostic value of tumor-infiltrating lymphocytes in Epstein-Barr virus-associated gastric cancer. Ann Oncol 2016; 27: 494-501. ArticlePubMedPDF
- 85. Cho J, Kang MS, Kim KM. Epstein-Barr virus-associated gastric carcinoma and specific features of the accompanying immune response. J Gastric Cancer 2016; 16: 1-7. ArticlePubMedPMCPDF
- 86. Derks S, Liao X, Chiaravalli AM, et al. Abundant PD-L1 expression in Epstein-Barr virus-infected gastric cancers. Oncotarget 2016; 7: 32925-32. ArticlePubMedPMC
- 87. Naseem M, Barzi A, Brezden-Masley C, et al. Outlooks on Epstein-Barr virus associated gastric cancer. Cancer Treat Rev 2018; 66: 15-22. ArticlePubMedPMC
- 88. Yau TO, Tang CM, Yu J. Epigenetic dysregulation in Epstein-Barr virus-associated gastric carcinoma: disease and treatments. World J Gastroenterol 2014; 20: 6448-56. ArticlePubMedPMC
- 89. Ambinder RF, Mann RB. Epstein-Barr-encoded RNA in situ hybridization: diagnostic applications. Hum Pathol 1994; 25: 602-5. ArticlePubMed
- 90. Gulley ML. Molecular diagnosis of Epstein-Barr virus-related diseases. J Mol Diagn 2001; 3: 1-10. ArticlePubMedPMC
- 91. Chen XZ, Chen H, Castro FA, Hu JK, Brenner H. Epstein-Barr virus infection and gastric cancer: a systematic review. Medicine (Baltimore) 2015; 94: e792.PubMedPMC
- 92. Melendez B, Van Campenhout C, Rorive S, Remmelink M, Salmon I, D’Haene N. Methods of measurement for tumor mutational burden in tumor tissue. Transl Lung Cancer Res 2018; 7: 661-7. ArticlePubMedPMC
- 93. Van Allen EM, Wagle N, Stojanov P, et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat Med 2014; 20: 682-8. ArticlePubMedPMCPDF
- 94. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015; 348: 124-8. PubMedPMC
- 95. Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014; 371: 2189-99. ArticlePubMedPMC
- 96. Hellmann MD, Ciuleanu TE, Pluzanski A, et al. Nivolumab plus Ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med 2018; 378: 2093-104. ArticlePubMedPMC
- 97. Steuer CE, Ramalingam SS. Tumor mutation burden: leading immunotherapy to the era of precision medicine? J Clin Oncol 2018; 36: 631-2. ArticlePubMed
- 98. Wang F, Wei XL, Wang FH, et al. Safety, efficacy and tumor mutational burden as a biomarker of overall survival benefit in chemorefractory gastric cancer treated with toripalimab, a PD-1 antibody in phase Ib/II clinical trial NCT02915432. Ann Oncol 2019; 30: 1479-86. ArticlePubMedPMCPDF
- 99. Griffith M, Miller CA, Griffith OL, et al. Optimizing cancer genome sequencing and analysis. Cell Syst 2015; 1: 210-23. ArticlePubMedPMC
- 100. Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 2017; 9: 34.ArticlePubMedPMCPDF
- 101. He Y, Rivard CJ, Rozeboom L, et al. Lymphocyte-activation gene-3, an important immune checkpoint in cancer. Cancer Sci 2016; 107: 1193-7. ArticlePubMedPMCPDF
- 102. Woo SR, Turnis ME, Goldberg MV, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res 2012; 72: 917-27. ArticlePubMedPDF
- 103. Coutzac C, Pernot S, Chaput N, Zaanan A. Immunotherapy in advanced gastric cancer, is it the future? Crit Rev Oncol Hematol 2019; 133: 25-32. ArticlePubMed
- 104. Zhu C, Anderson AC, Schubart A, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol 2005; 6: 1245-52. ArticlePubMedPDF
- 105. Yanai H, Ban T, Wang Z, et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature 2009; 462: 99-103. ArticlePubMedPDF
- 106. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med 2010; 207: 2187-94. ArticlePubMedPMCPDF
- 107. Prendergast GC, Smith C, Thomas S, et al. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother 2014; 63: 721-35. ArticlePubMedPMCPDF
- 108. Hornyak L, Dobos N, Koncz G, et al. The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front Immunol 2018; 9: 151.PubMedPMC
- 109. Terness P, Bauer TM, Röse L, et al. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med 2002; 196: 447-57. PubMedPMCPDF
- 110. Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer 2014; 2: 3.ArticlePubMedPMC
- 111. Zaretsky JM, Garcia-Diaz A, Shin DS, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med 2016; 375: 819-29. PubMedPMC
- 112. Chowell D, Morris LG, Grigg CM, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2018; 359: 582-7. PubMed
REFERENCES
Figure & Data
References
Citations
- Using glucocorticoid receptor-related genes to create and validate a survival model predicting gastric cancer
Ke Guo, Ping Huang, Jiasheng Zhang, Bo Zhang, Linyang Li, Jiaxin Li
Computational Biology and Chemistry.2026; 120: 108726. CrossRef - Mechanisms of Metabolic Reprogramming Regulating Immunosuppression in the Gastric Cancer Tumor Microenvironment
Wenting Dong, Xuepeng Qian, Honglin Liu, Jinhai Huo, Weiming Wang
Biomolecules.2026; 16(1): 160. CrossRef - Perineural invasion in digestive tract tumors: Immune system interactions and therapeutic strategies
Jia-Yu Pang, Rong-Yao Jin, Han-Xiao Zhang, Ying-Hua Zhang, Xin-Yue Wei, Wen-Bo Cao, Yi-Xiao Chen, Jun-Ling Wang, Sai-Jun Mo
World Journal of Clinical Oncology.2026;[Epub] CrossRef - FOXP3+/CD8+ ratio associated with aggressive behavior in RUNX3‐methylated diffuse esophagogastric junction tumor
Suguru Maruyama, Yu Imamura, Tasuku Toihata, Ikumi Haraguchi, Manabu Takamatsu, Makiko Yamashita, Yuichiro Nakashima, Eiji Oki, Kenichi Taguchi, Manabu Yamamoto, Shinji Mine, Akihiko Okamura, Jun Kanamori, Souya Nunobe, Takeshi Sano, Shigehisa Kitano, Tet
Cancer Science.2025; 116(1): 178. CrossRef - Immune biomarkers and predictive signatures in gastric cancer: Optimizing immunotherapy responses
Sundaram Vickram, Shofia Saghya Infant, S. Manikandan, D. Jenila Rani, C.M. Mathan Muthu, Hitesh Chopra
Pathology - Research and Practice.2025; 265: 155743. CrossRef - Korean Practice Guidelines for Gastric Cancer 2024: An Evidence-based, Multidisciplinary Approach (Update of 2022 Guideline)
In-Ho Kim, Seung Joo Kang, Wonyoung Choi, An Na Seo, Bang Wool Eom, Beodeul Kang, Bum Jun Kim, Byung-Hoon Min, Chung Hyun Tae, Chang In Choi, Choong-kun Lee, Ho Jung An, Hwa Kyung Byun, Hyeon-Su Im, Hyung-Don Kim, Jang Ho Cho, Kyoungjune Pak, Jae-Joon Kim
Journal of Gastric Cancer.2025; 25(1): 5. CrossRef - Targeted therapy and immunotherapy for gastric cancer: rational strategies, novel advancements, challenges, and future perspectives
Dong Luo, Yunmei Liu, Zhengmao Lu, Lei Huang
Molecular Medicine.2025;[Epub] CrossRef - Prognostic value of the triglyceride-glucose index in gastric cancer
Tugce Eskazan, Suat Saribas, Bekir Kocazeybek
World Journal of Gastroenterology.2025;[Epub] CrossRef - Deciphering the dual role of autophagy in gastric cancer and gastroesophageal junction cancer: from tumor suppression to cancer progression
Lili Lei, Junling Zhang, Ran Wei, Bingqi Dong, Xin Wang, Ying Zhou
Discover Oncology.2025;[Epub] CrossRef - Comparison of clinicopathological parameters with the presence of Epstein–Barr virus and the absence of DNA mismatch repair proteins in gastric adenocarcinomas
Özge Eyeoğlu, Serra Kayaçetin
Asian Biomedicine.2025; 19(2): 86. CrossRef - The impact of combined immunotherapy on the cellular composition of the tumor microenvironment in patients with gastric carcinoma
L.A. Tashireva, A.Yu. Kalinchuk, D.M. Loos, E.A. Tsarenkova, A.V. Avgustinovich, S.G. Afanas’ev, S.V. Vtorushin
Russian Journal of Archive of Pathology.2025; 87(4): 24. CrossRef - Immunotherapy in gastric adenocarcinoma – a rapidly evolving treatment landscape
Yang Wang, Geoffrey Chong
Critical Reviews in Oncology/Hematology.2025; 216: 104941. CrossRef - Ubiquilin-4 induces immune escape in gastric cancer by activating the notch signaling pathway
Quan Jiang, Hao Chen, Shixin Zhou, Tao Zhu, Wenshuai Liu, Hao Wu, Yong Zhang, Fenglin Liu, Yihong Sun
Cellular Oncology.2024; 47(1): 303. CrossRef - Expression and prognostic value of APOBEC2 in gastric adenocarcinoma and its association with tumor-infiltrating immune cells
Lipan Wei, Xiuqian Wu, Lan Wang, Ling Chen, Xuejun Wu, Tiantian Song, Yuanyuan Wang, Wenjun Chang, Aizhen Guo, Yongdong Niu, Haihua Huang
BMC Cancer.2024;[Epub] CrossRef - Identification and characterization of CLEC11A and its derived immune signature in gastric cancer
Qing Zheng, Zhenqi Gong, Baizhi Li, Runzi Cheng, Weican Luo, Cong Huang, Huaiming Wang
Frontiers in Immunology.2024;[Epub] CrossRef - Cervical cancer subtype identification and model building based on lipid metabolism and post-infection microenvironment immune landscape
Yongzhi Chen, Rongjie Cui, Dun Xiong, Yuan Zhao, Jianyu Pang, Samina Gul, Qi Qi, Yuheng Tang, Xuhong Zhou, Wenru Tang
Heliyon.2024; 10(9): e30746. CrossRef - Systematic Analysis of Tumor Stem Cell-related Gene Characteristics
to Predict the PD-L1 Immunotherapy and Prognosis of Gastric
Cancer
Chenchen Wang, Ying Chen, Ru Zhou, Ya’nan Yang, Yantian Fang
Current Medicinal Chemistry.2024; 31(17): 2467. CrossRef - Comprehensive landscape of m6A regulator-related gene patterns and tumor microenvironment infiltration characterization in gastric cancer
Bin Peng, Yinglin Lin, Gao Yi, Mingzhen Lin, Yao Xiao, Yezhenghong Qiu, Wenxia Yao, Xinke Zhou, Zhaoyu Liu
Scientific Reports.2024;[Epub] CrossRef - Distinctive Phenotypic and Microenvironmental Characteristics of Neuroendocrine Carcinoma and Adenocarcinoma Components in Gastric Mixed Adenoneuroendocrine Carcinoma
Yoonjin Kwak, Soo Kyung Nam, Yujun Park, Yun-Suhk Suh, Sang-Hoon Ahn, Seong-Ho Kong, Do Joong Park, Hyuk-Joon Lee, Hyung-Ho Kim, Han-Kwang Yang, Hye Seung Lee
Modern Pathology.2024; 37(10): 100568. CrossRef - Computed tomography-detected extramural venous invasion-related gene signature: a potential negative biomarker of immune checkpoint inhibitor treatment in patients with gastric cancer
Hao Yang, Xinyi Gou, Caizhen Feng, Yinli Zhang, Fan Chai, Nan Hong, Yingjiang Ye, Yi Wang, Bo Gao, Jin Cheng
Journal of Translational Medicine.2023;[Epub] CrossRef - A standardized pathology report for gastric cancer: 2nd edition
Young Soo Park, Myeong-Cherl Kook, Baek-hui Kim, Hye Seung Lee, Dong-Wook Kang, Mi-Jin Gu, Ok Ran Shin, Younghee Choi, Wonae Lee, Hyunki Kim, In Hye Song, Kyoung-Mee Kim, Hee Sung Kim, Guhyun Kang, Do Youn Park, So-Young Jin, Joon Mee Kim, Yoon Jung Choi,
Journal of Pathology and Translational Medicine.2023; 57(1): 1. CrossRef - A Standardized Pathology Report for Gastric Cancer: 2nd Edition
Young Soo Park, Myeong-Cherl Kook, Baek-hui Kim, Hye Seung Lee, Dong-Wook Kang, Mi-Jin Gu, Ok Ran Shin, Younghee Choi, Wonae Lee, Hyunki Kim, In Hye Song, Kyoung-Mee Kim, Hee Sung Kim, Guhyun Kang, Do Youn Park, So-Young Jin, Joon Mee Kim, Yoon Jung Choi,
Journal of Gastric Cancer.2023; 23(1): 107. CrossRef - Korean Practice Guidelines for Gastric Cancer 2022: An Evidence-based, Multidisciplinary Approach
Tae-Han Kim, In-Ho Kim, Seung Joo Kang, Miyoung Choi, Baek-Hui Kim, Bang Wool Eom, Bum Jun Kim, Byung-Hoon Min, Chang In Choi, Cheol Min Shin, Chung Hyun Tae, Chung sik Gong, Dong Jin Kim, Arthur Eung-Hyuck Cho, Eun Jeong Gong, Geum Jong Song, Hyeon-Su Im
Journal of Gastric Cancer.2023; 23(1): 3. CrossRef - Could Toll-like Receptor 2 Serve as Biomarker to Detect Advanced Gastric Cancer?
Marek Majewski, Kamil Torres, Paulina Mertowska, Sebastian Mertowski, Izabela Korona-Głowniak, Jan Korulczyk, Witold Zgodziński, Ewelina Grywalska
International Journal of Molecular Sciences.2023; 24(6): 5824. CrossRef - Research Progress of Immunotherapy for Gastric Cancer
Zhipeng Zhang, Ningning Liu, Mingyu Sun
Technology in Cancer Research & Treatment.2023;[Epub] CrossRef - Case Report: A rare synchronous multiple gastric carcinoma achieved progression-free disease through NGS-guided serial treatment
Xinyi Shao, Jin Yin, Di Wang, Erjiong Huang, Yini Zhang, Jiani C. Yin, Chen Huang, Hao Wu, Xiaoli Wu
Frontiers in Oncology.2023;[Epub] CrossRef - Artificial Intelligence-Enabled Gastric Cancer Interpretations
Mustafa Yousif, Liron Pantanowitz
Surgical Pathology Clinics.2023; 16(4): 673. CrossRef - The Optimal Tumor Mutational Burden Cutoff Value as a Novel Marker for Predicting the Efficacy of Programmed Cell Death-1 Checkpoint Inhibitors in Advanced Gastric Cancer
Jae Yeon Jang, Youngkyung Jeon, Sun Young Jeong, Sung Hee Lim, Won Ki Kang, Jeeyun Lee, Seung Tae Kim
Journal of Gastric Cancer.2023; 23(3): 476. CrossRef - Biomarkers for Predicting Response to Personalized Immunotherapy in Gastric Cancer
Moonsik Kim, Ji Yun Jeong, An Na Seo
Diagnostics.2023; 13(17): 2782. CrossRef - The Prognostic Value of the Prognostic Nutritional Index in Patients with Advanced or Metastatic Gastric Cancer Treated with Immunotherapy
Yuting Pan, Yue Ma, Guanghai Dai
Nutrients.2023; 15(19): 4290. CrossRef - Molecular classification of gastric cancer predicts survival in patients undergoing radical gastrectomy based on project HOPE
Kenichiro Furukawa, Keiichi Hatakeyama, Masanori Terashima, Takeshi Nagashima, Kenichi Urakami, Keiichi Ohshima, Akifumi Notsu, Takashi Sugino, Taisuke Yagi, Keiichi Fujiya, Satoshi Kamiya, Makoto Hikage, Yutaka Tanizawa, Etsuro Bando, Yae Kanai, Yasuto A
Gastric Cancer.2022; 25(1): 138. CrossRef - Immunotherapy for Gastric Cancer: A 2021 Update
Christo Kole, Nikolaos Charalampakis, Sergios Tsakatikas, Nikolaos-Iasonas Kouris, George Papaxoinis, Michalis V Karamouzis, Anna Koumarianou, Dimitrios Schizas
Immunotherapy.2022; 14(1): 41. CrossRef - The immune microenvironment in gastric adenocarcinoma
Yana Zavros, Juanita L. Merchant
Nature Reviews Gastroenterology & Hepatology.2022; 19(7): 451. CrossRef - Immunomodulation by Gut Microbiome on Gastrointestinal Cancers: Focusing on Colorectal Cancer
Raghad Khalid AL-Ishaq, Lenka Koklesova, Peter Kubatka, Dietrich Büsselberg
Cancers.2022; 14(9): 2140. CrossRef - An Immunity-Associated lncRNA Signature for Predicting Prognosis in Gastric Adenocarcinoma
Xiaowen Zhao, Pingfan Wu, Dongling Liu, Changtian Li, Ling Xue, Zhe Liu, Meng Zhu, Jie Yang, Ziyi Chen, Yaling Li, Yali She, Kathiravan Srinivasan
Journal of Healthcare Engineering.2022; 2022: 1. CrossRef - RNA modification writers influence tumor microenvironment in gastric cancer and prospects of targeted drug therapy
Peng Song, Sheng Zhou, Xiaoyang Qi, Yuwen Jiao, Yu Gong, Jie Zhao, Haojun Yang, Zhifen Qian, Jun Qian, Liming Tang
Journal of Bioinformatics and Computational Biology.2022;[Epub] CrossRef - Identification of the three subtypes and the prognostic characteristics of stomach adenocarcinoma: analysis of the hypoxia-related long non-coding RNAs
Zehua Fan, Yanqun Wang, Rong Niu
Functional & Integrative Genomics.2022; 22(5): 919. CrossRef - Complete Response of High Microsatellite Instability Gastric Cancer and Synchronous Microsatellite Stability Rectal Cancer
Zachary E Hunzeker, Pooja Bhakta, Sindusha R Gudipally, Sri Bharathi Kavuri, Rohit Venkatesan, Chukwuyejulumafor Nwanze
Cureus.2022;[Epub] CrossRef - Immune Profiling in Gastric Cancer Reveals the Dynamic Landscape of Immune Signature Underlying Tumor Progression
Yuhan Wei, Jianwei Zhang, Xueke Fan, Zhi Zheng, Xiaoyue Jiang, Dexi Chen, Yuting Lu, Yingrui Li, Miao Wang, Min Hu, Qi Du, Liuting Yang, Hongzhong Li, Yi Xiao, Yongfu Li, Jiangtao Jin, Deying Wang, Xiangliang Yuan, Qin Li
Frontiers in Immunology.2022;[Epub] CrossRef - Tumor vessel normalization and immunotherapy in gastric cancer
Xianzhe Yu, Shan He, Jian Shen, Qiushi Huang, Peng Yang, Lin Huang, Dan Pu, Li Wang, Lu Li, Jinghua Liu, Zelong Liu, Lingling Zhu
Therapeutic Advances in Medical Oncology.2022;[Epub] CrossRef - FN1 is a prognostic biomarker and correlated with immune infiltrates in gastric cancers
Han Wang, Junchang Zhang, Huan Li, Hong Yu, Songyao Chen, Shuhao Liu, Changhua Zhang, Yulong He
Frontiers in Oncology.2022;[Epub] CrossRef - Molecular Pathology of Gastric Cancer
Moonsik Kim, An Na Seo
Journal of Gastric Cancer.2022; 22(4): 264. CrossRef - Bioinformatics Analysis and Structure of Gastric Cancer Prognosis Model Based on Lipid Metabolism and Immune Microenvironment
Yongzhi Chen, Hongjun Yuan, Qian Yu, Jianyu Pang, Miaomiao Sheng, Wenru Tang
Genes.2022; 13(9): 1581. CrossRef - Clinical implications of interleukins-31, 32, and 33 in gastric cancer
Qing-Hua Liu, Ji-Wei Zhang, Lei Xia, Steven G Wise, Brett David Hambly, Kun Tao, Shi-San Bao
World Journal of Gastrointestinal Oncology.2022; 14(9): 1808. CrossRef - Microbiota and the Immune System—Actors in the Gastric Cancer Story
Marek Majewski, Paulina Mertowska, Sebastian Mertowski, Konrad Smolak, Ewelina Grywalska, Kamil Torres
Cancers.2022; 14(15): 3832. CrossRef - Bioinformatics and Experimental Analyses Reveal MAP4K4 as a Potential Marker for Gastric Cancer
Junping Zhang, Xiaoping Cai, Weifeng Cui, Zheng Wei
Genes.2022; 13(10): 1786. CrossRef - Common strategies for effective immunotherapy of gastroesophageal cancers using immune checkpoint inhibitors
Shuang Ma, Fei Chen
Pathology - Research and Practice.2022; 238: 154110. CrossRef - High-level of intratumoral GITR+ CD4 T cells associate with poor prognosis in gastric cancer
Shouyu Ke, Feng Xie, Yixian Guo, Jieqiong Chen, Zeyu Wang, Yimeng Yu, Haigang Geng, Danhua Xu, Xu Liu, Xiang Xia, Fengrong Yu, Chunchao Zhu, Zizhen Zhang, Gang Zhao, Bin Li, Wenyi Zhao
iScience.2022; 25(12): 105529. CrossRef - Characteristics of Adenosine-to-Inosine RNA editing-based subtypes and novel risk score for the prognosis and drug sensitivity in stomach adenocarcinoma
Jingjing Pan, Xinyuan Gu, Jing Luo, Xinye Qian, Qiang Gao, Tianjie Li, Longying Ye, Chenlu Li
Frontiers in Cell and Developmental Biology.2022;[Epub] CrossRef - Inhibition of NF‐κB is required for oleanolic acid to downregulate PD‐L1 by promoting DNA demethylation in gastric cancer cells
Xirong Lu, Yuyi Li, Wei Yang, Minghao Tao, Yanmiao Dai, Jinkang Xu, Qianfei Xu
Journal of Biochemical and Molecular Toxicology.2021;[Epub] CrossRef - Prognostic Value of C-Reactive Protein to Albumin Ratio in Gastric Cancer: A Meta-Analysis
Liang Yue, Yi Lu, Yulin Li, Yilin Wang
Nutrition and Cancer.2021; 73(10): 1864. CrossRef - Immunogenic characteristics of microsatellite instability‐low esophagogastric junction adenocarcinoma based on clinicopathological, molecular, immunological and survival analyses
Yu Imamura, Tasuku Toihata, Ikumi Haraguchi, Yoko Ogata, Manabu Takamatsu, Aya Kuchiba, Norio Tanaka, Osamu Gotoh, Seiichi Mori, Yuichiro Nakashima, Eiji Oki, Masaki Mori, Yoshinao Oda, Kenichi Taguchi, Manabu Yamamoto, Masaru Morita, Naoya Yoshida, Hideo
International Journal of Cancer.2021; 148(5): 1260. CrossRef - Two Similar Signatures for Predicting the Prognosis and Immunotherapy Efficacy of Stomach Adenocarcinoma Patients
Taohua Yue, Shuai Zuo, Jing Zhu, Shihao Guo, Zhihao Huang, Jichang Li, Xin Wang, Yucun Liu, Shanwen Chen, Pengyuan Wang
Frontiers in Cell and Developmental Biology.2021;[Epub] CrossRef - Tumor microenvironment characterization in stage IV gastric cancer
Feng Yang, Zhenbao Wang, Xianxue Zhang
Bioscience Reports.2021;[Epub] CrossRef - E2F2 inhibition induces autophagy via the PI3K/Akt/mTOR pathway in gastric cancer
Hui Li, Shufen Zhao, Liwei Shen, Peige Wang, Shihai Liu, Yingji Ma, Zhiwei Liang, Gongjun Wang, Jing Lv, Wensheng Qiu
Aging.2021; 13(10): 13626. CrossRef - Chemoradiation induces upregulation of immunogenic cell death-related molecules together with increased expression of PD-L1 and galectin-9 in gastric cancer
S. H. Petersen, L. F. Kua, S. Nakajima, W. P. Yong, K. Kono
Scientific Reports.2021;[Epub] CrossRef - Establishment of an Immune Cell Infiltration Score to Help Predict the Prognosis and Chemotherapy Responsiveness of Gastric Cancer Patients
Quan Jiang, Jie Sun, Hao Chen, Chen Ding, Zhaoqing Tang, Yuanyuan Ruan, Fenglin Liu, Yihong Sun
Frontiers in Oncology.2021;[Epub] CrossRef - Microsatellite instability in Gastric Cancer: Between lights and shadows
Elisabetta Puliga, Simona Corso, Filippo Pietrantonio, Silvia Giordano
Cancer Treatment Reviews.2021; 95: 102175. CrossRef - Survival-associated alternative splicing events interact with the immune microenvironment in stomach adenocarcinoma
Zai-Sheng Ye, Miao Zheng, Qin-Ying Liu, Yi Zeng, Sheng-Hong Wei, Yi Wang, Zhi-Tao Lin, Chen Shu, Qiu-Hong Zheng, Lu-Chuan Chen
World Journal of Gastroenterology.2021; 27(21): 2871. CrossRef - Immunotherapy of gastric cancer: Past, future perspective and challenges
Jun Xie, Liping Fu, Li Jin
Pathology - Research and Practice.2021; 218: 153322. CrossRef - Clinicopathologic and Prognostic Association of GRP94 Expression in Colorectal Cancer with Synchronous and Metachronous Metastases
Sumi Yun, Sukmook Lee, Ho-Young Lee, Hyeon Jeong Oh, Yoonjin Kwak, Hye Seung Lee
International Journal of Molecular Sciences.2021; 22(13): 7042. CrossRef - Injectable shear-thinning polylysine hydrogels for localized immunotherapy of gastric cancer through repolarization of tumor-associated macrophages
Yan Yang, Yang Yang, Meili Chen, Jianquan Chen, Jinyan Wang, Yajun Ma, Hanqing Qian
Biomaterials Science.2021; 9(19): 6597. CrossRef - Correlation between LRP1B Mutations and Tumor Mutation Burden in Gastric Cancer
Sizhe Hu, Xiaokang Zhao, Feng Qian, Cancan Jin, Kaishun Hou, Tao Huang
Computational and Mathematical Methods in Medicine.2021; 2021: 1. CrossRef - Comprehensive Analysis to Identify MAGEA3 Expression Correlated With Immune Infiltrates and Lymph Node Metastasis in Gastric Cancer
Jinji Jin, Jianxin Tu, Jiahuan Ren, Yiqi Cai, Wenjing Chen, Lifang Zhang, Qiyu Zhang, Guanbao Zhu
Frontiers in Oncology.2021;[Epub] CrossRef - Effect of P2X7 receptor on tumorigenesis and its pharmacological properties
Wen-jun Zhang, Ce-gui Hu, Zheng-ming Zhu, Hong-liang Luo
Biomedicine & Pharmacotherapy.2020; 125: 109844. CrossRef - Current status and future potential of predictive biomarkers for immune checkpoint inhibitors in gastric cancer
Byung Woog Kang, Ian Chau
ESMO Open.2020; 5(4): e000791. CrossRef - Is Ramucirumab Still the Only Second-Line Treatment in Metastatic Gastric Cancer?
Khalil El Gharib, Hampig Raphael Kourie
Pharmacogenomics.2020; 21(17): 1203. CrossRef - Deep Learning Predicts Underlying Features on Pathology Images with Therapeutic Relevance for Breast and Gastric Cancer
Renan Valieris, Lucas Amaro, Cynthia Aparecida Bueno de Toledo Osório, Adriana Passos Bueno, Rafael Andres Rosales Mitrowsky, Dirce Maria Carraro, Diana Noronha Nunes, Emmanuel Dias-Neto, Israel Tojal da Silva
Cancers.2020; 12(12): 3687. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
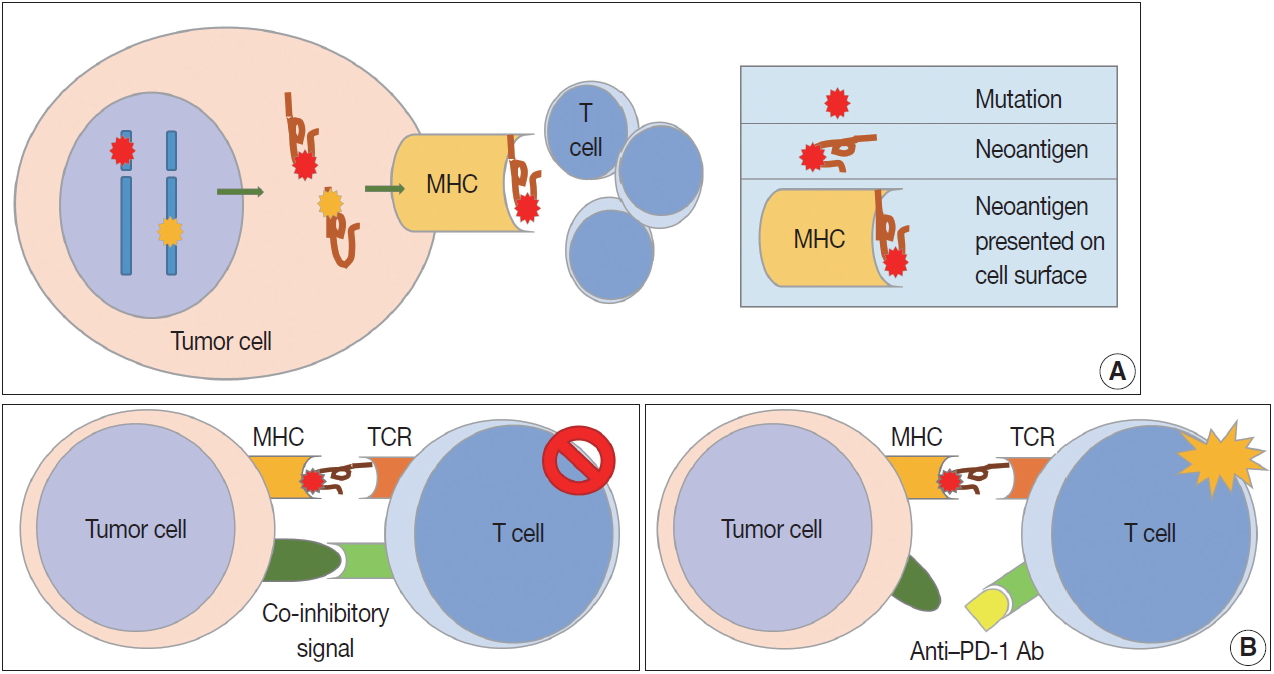
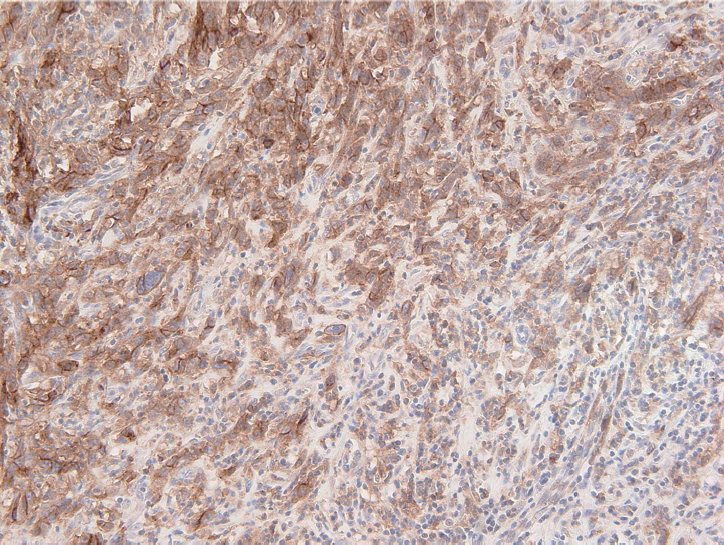
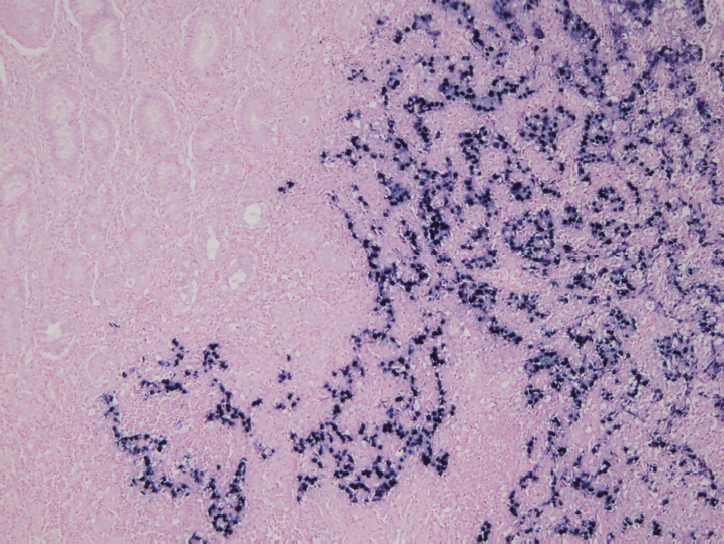



Fig. 1.
Fig. 2.
Fig. 3.
| Study | Region | No. | Subsets | Outcomes | TMA | Study | Selected area | CD8 cutoff point | CD8 cutoff number (/mm2) |
|---|---|---|---|---|---|---|---|---|---|
| Lee et al. (2008) [29] | Korea | 220 | CD3/CD8/CD45RO | OS | Yes | Consecutive GC | Representative one area | Mean | 435.73 |
| Haas et al. (2009) [30] | Germany | 52 | CD3/CD8/CD20/Foxp3/Granzyme B/M | OS | Yes | Gastric cardia cancer | Six representative areas | Median | 21.6 (epithelial) |
| 212.7 (stromal) | |||||||||
| Shen et al. (2010) [31] | China | 133 | CD4/CD8/Foxp3 | OS | Yes | GC with R0 resection | Average of two centers and two invasive border | Median | 946.22 (intratumoral) |
| 744.40 (peritumoral) | |||||||||
| Kim et al. (2011) [32] | Korea | 180 | CD3/CD4/CD8/Foxp3/Granzyme B | OS/RFS | No | Gastric cardia cancer | Mean of 5 HPFs (center areas) | Median | 60.8/HPF (253.33) |
| Kim et al. (2014) [33] | Korea | 99 | CD8/Foxp3 | OS | Yes | MSI-H advanced GC | Average of 3 representative areas | 60th percentile | 601.5 (median, 542.6) |
| Li e al. (2015) [34] | China | 192 | CD4/CD8 | OS | No | Advanced GC | Representative one slide | 26%-100% staining | Not available |
| Liu et al. (2015) [35] | China | 166 | CD3/CD4/CD8/Foxp3/CD57/M | OS | No | Surgical resection cases | Average of 5 noncontiguous and the densest random areas, in intratumoral and stromal area | Median | 839.69 (intratumoral) |
| 523.90 (stromal) | |||||||||
| Hennequin et al. (2016) [36] | France | 82 | CD8/CD20/Foxp3/Tbet | RFS | No | Consecutive GC (including preop-chemotherapy) | Mean of 3 HPF in core and invasive margin | Median | Not available |
| Kim et al. (2016) [37] | Korea | 243 | CD3/CD4/CD8 | DFS | Yes | Consecutive GC | Representative one core | Median | 375.48 |
| Giampieri et al. (2017) [38] | Italy | 73 | CD3 | OS | No | Metastatic GC with 1st-line chemotherapy | Biopsy or resected specimens | More than 50%-60% stromal area covered by TILs | Not available |
| Kawazoe et al. (2017) [39] | Japan | 383 | CD3/CD4/CD8/Foxp3 | OS | Yes | Advanced GC | Invasive area (two cores) | Median | 384 |
| Koh et al. (2017) [40] | Korea | 392 | CD3/CD4/CD8/Foxp3 | OS | Yes | Stage II and III GC | Tumor center and invasive border | 25th percentile | 130.07 (center) |
| 101.76 (border) | |||||||||
| Pernot et al. (2019) [41] | France | 67 | CD8/Foxp3/CD57 | OS | No | Locally advanced or metastatic GC | Mean of 3 representative HPFs | Median | 31/HPF |
| Kim et al. (2019) [42] | Korea | 297 | CD3/CD8/Foxp3 | OS | Yes | Early GC with submucosal invasion and advanced GC | Tumor center and invasive border | Median | Not available |
| Trials | Target | Phase | Treatment arms | Setting (line) | No. of patients | Results/primary endpoints | |
|---|---|---|---|---|---|---|---|
| Pembralizumab | |||||||
| KEYNOTE-012 [49] | PD-1 | 1b | Pembrolizumab | Any | 39 with positive PD-L1 | ORR (%): 22 (95% CI, 10-39; all PR) | |
| KEYNOTE-059 (cohort 1) [50] | PD-1 | 2 | Pembrolizumab | ≥ 3rd | 259 | ORR (%): 11.6 (95% CI, 8.0–16.1; CR in 2.3) | |
| Median response duration (mo): 8.4 (1.6 + |
|||||||
| OS (mo): 5.6; PFS (mo): 2.0 | |||||||
| KEYNOTE-061 [51] | PD-1 | 3 | Pembrolizumab | 2nd | 592 (395 with CPS ≥ 1) | OS (mo): 9.1 vs. 8.3 (HR, 0.82; 95% CI, 0.66-1.03) | |
| PaCIitaxel | PFS (mo): 1.5 vs. 4.1 (HR, 1.27; 95% CI, 1.03-1.57) (both analyses in the subgroup of positive PD-L1) | ||||||
| KEYNOTE-062 [52] | PD-1 | 3 | Pembrolizumab vs | 1st | 763 with CPS ≥ 1 (281 with CPS ≥ 10) | OS (mo): 10.6 vs. 12.5 vs. 11.1 (CPS ≥ 1) | |
| Pembrolizumab + cisplatin + 5-FU or capecitabine | OS (mo): 17.4 vs. 12.3 vs. 10.8 (CPS ≥ 10) | ||||||
| PFS (mo): 2.0 vs. 6.9 vs. 6.4 (CPS ≥ 1) | |||||||
| Placebo + cisplatin + 5-FU or capecitabine | PFS (mo): 2.9 vs. 5.7 vs. 6.1 (CPS ≥ 10) | ||||||
| Nivolumab (± ipilimumab) | |||||||
| CheckMate-032 [53] | PD-1 | 1/2 | Nivolumab 3 mg/kg nivolumab 1 mg/kg + ipilimumab 3 mg/kg | ≥ 2nd | 160 | ORR (%): 12 vs. 24 vs. 8 (independent of PD-L1 status) | |
| CTLA-4 | |||||||
| Nivolumab 3 mg/kg+ipilimumab 1 mg/kg | 12-mo PFS rates (%): 8 vs. 17 vs. 10 | ||||||
| 12-mo OS rates (%): 39 vs. 35 vs. 24 | |||||||
| ONO-4538-12, ATTRACTION-2 [46] | PD-1 | 3 | Nivolumab alone placebo | ≥ 3rd | 493 | OS (mo): 5.26 vs. 4.14 (HR, 0.63; 95% CI, 0.51-0.78) | |
| 12-mo OS rates (%): 26.2 vs. 10.9 | |||||||
| ATTRACTION-4, part 1 [54] | PD-1 | 2 | Nivolumab + oxaliplatin+capecitabine | 1st | 40 | ORR (%): 76.5 vs. 57.1 | |
| Nivdumab+oxaliplatin+S-1 | PFS (mo): 10.6 vs. 9.7 | ||||||
| ATTRACTION-4, part 2 | PD-1 | 3 | Nivolumab + oxaliplatin+S-1 or capecitabine Placebo + oxaliplatin+S-1 or capecitabine | 1st | Approx. 650 | Ongoing | |
| CheckMate-649 [55] | PD-1 | 3 | Nivolumab + ipilimumab | 1st | 870 | Ongoing | |
| CTLA-4 | Nivolumab + oxaliplatin + 5-FU or capecitabine Oxaliplatin + 5-FU or capecitabine | ||||||
| Others | |||||||
| JAVELIN Gastric 100 [58] | PD-L1 | 3 | Avelumab | Maintenance after 1st-line | 499 | Ongoing | |
| BSC after response or stability to oxaliplatinb + fluoropyrimidine | |||||||
| JAVELIN Gastric 300 [57] | PD-L1 | 3 | Avelumab | 3rd | 371 | OS (mo): 4.6 vs. 5.0 (HR, 1.1; 95% CI, 0.9-1.4) | |
| PaCIitaxel or irinotecan | PFS (mo): 1.4 vs. 2.7 (HR, 1.73; 95% CI, 1.4-2.2) | ||||||
| ORR (%): 2.2 vs. 4.3 | |||||||
| NCT02340975 [59] | PD-L1 | 1b/2 | Durvalumab (anti-PD-L1) | ≥ 2nd | 94 (phase 2; as of Sep 13, 2017) | Ongoing | |
| CTLA-4 | Tremelimumab (anti-CTLA-4) | ||||||
| Durvalumab+tremelimumab | |||||||
| NCT01968109 [60] | LAG-3 | 1/2a | Relatlimab (anti-LAG3) vs. Relatlimab + nivolumab | Last | Advanced solid tumors | Ongoing | |
| PD-1 | |||||||
TMA, tissue microarray; OS, overall survival; GC, gastric cancer; RFS, relapse-free survival; HPF, high power field; MSI-H, microsatellite instability-high; DFS, disease-free survival; TIL, tumor-infiltrating lymphocyte.
PD-1, programmed death-1; PD-L1, programmed death-ligand 1; ORR, objective response rate; CI, confidence interval; PR, partial response; CR, complete response; OS, overall survival; PFS, progression-free survival; CPS, Combined Positive Score for PD-L1; HR, hazard ratio; CI, confidence interval; 5-FU, 5-fluorouracil; CTLA-4, cytotoxic T lymphocyte–associated protein 4; LAG-3, lymphocyte-activation gene 3. + indicates that patients had no progressive disease at their last assessment.

